Part 1: Gene function
How are genes identified? What is a gene?
Usually, one of the first steps upon identifying all of the nucleotide sequences in a genome is to identify all potential genes that may exist in the genome. Typically, this is completed using a computer program that runs a variety of mathematical algorithms to identify potential genes. However, to better understand this process, it is important to identify the critical components of a gene. Therefore, in this exercise, we will use a small portion of a sequenced genome to identify a gene.
We will be working with a bacterial chromosome. Bacterial chromosomes are commonly constructed as a single circular double-stranded piece of DNA. Their size can range from just under one to 6 million nucleotides.
Before starting this exercise, update yourself on gene definition (in bioinformatics context, gene structure, and gene function) and formulate an answer to a superior easy question: what role does gene play in the central dogma (the process of transcription and translation)?
Imagine that a newly identified bacterium has just had its genome sequenced. Propose how you might identify a gene in its genome (i.e., what will you look for in the sequence to characterize a gene?).
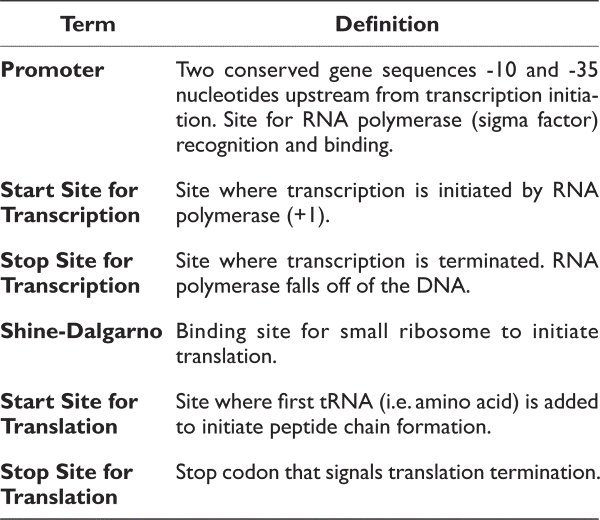
The conserved components of all genes are the start and stop codons for translation. Therefore, the first step should be identifying the start and stop codons to predict where protein borders are. Then, it would be essential to identify promoters and Shine-Dalgarno sites to verify the existence of the predicted gene.
Please note that promoters and Shine-Dalgarno sequences, although relatively conserved within an individual bacterium, vary from organism to organism. In addition, not all genes will have a promoter directly in front of the gene in an operon. Therefore, although these sequences are necessary, they are not commonly used for the initial identification of genes.
How might gene structure be similar or different when looking for genes in bacteria and eukaryotes?
Differences: Promoters are different in bacteria and eukaryotes. Eukaryotes contain a single sequence promoter (-35). This is because they use structurally different RNA polymerases and transcription factors responsible for binding to the promoter. Eukaryotes do not have a Shine-Dalgarno for ribosome binding. Instead, eukaryotes use the 5’ cap for ribosome binding.
Similarities: Start and stop sites for translation are similar. The structural location of gene components (i.e., the promoter, site for transcription, and translation starts and stops) is similar.
Below is a single strand of DNA taken from a bacterial chromosome. Identify the pieces listed below and answer the following questions to help you find the gene in this sequence. This strand represents the coding strand, meaning this is the strand that would be read during translation.
ctcattaggcaccccaggctttacactttatgcttccggctcgtatgttgtgtggaattgtgagcggataacaatttcaca
cacaaggaaacagctatgaccatcattacggattcactggccgtcgacggcaggccacgttcggcaatttaacg
agcgttattgaaataggcgggggcacgccccctctagtactcataaaaaaagtgatcat
The items below are all components of a gene. Please define each of these items. For example, when asked to define the start site for transcription, please identify what occurs at this location. In addition, please identify and label each region on the sequence above:
- Promoter (promoter regions are rich in Ts and As)
- Pribnow or -10 region: tatgtt
- start site for transcription (+1; figure out from promoter)
- start site for translation (atg)
- stop site for translation taa, tag or tga
- Shine-Dalgarno site (agga)
- Stop site for transcription termination (this will be an approximation)
Please feel free to find your way to present the results in the most informative way. First, I would import the sequence in GeneDoc, make the font large (25-30), and remove the consensus line (use Ctrl+G for the setup options). Then I would use one of the multiple web tools (or MEGA) to translate the nt sequence to aa (for example, ExPASy is very handy). So, you may immediately get an idea of whether there is a meaningful reading frame in this sequence. Then I would use the search tool in GeneDoc and search for essential motives. Finally, I would also use a snipping tool to copy the view from GeneDoc and paste it in, for example, PowerPoint for annotation. However, once again, you are skillful in finding how to present these relatively simple data.
What is the primary protein structure encoded by this gene? Remember, this strand represents the same sequence as the RNA copied during transcription. Although ideally, I would expect you to use the genetic code and do the manual translation of codons to amino acids, it should be practiced manually at least once. Replace the T’s with U’s to use the triple codon table. Of course, you may do the translation with one of the numerous web-based translating tools. Maybe you would wish to do both and compare the outcome.
You should have all of the terms mapped on the sequence. The order of these terms is essential for the successful transcription and translation of a gene. To address its importance, please answer the following questions regarding the gene’s structure:
- Where are the start and stop sites for translation concerning the start and stop site for transcription? Why is this important?
- Where is the Shine-Dalgarno site related to starting sites for transcription and translation? Why is this important?
- Where is the promoter related to the start sites for transcription and translation? Why is this important?
Please note that this sequence has been composed for the exercise (the polypeptide length has been truncated). However, if you are curious enough, you may use the sequence similarity search and identify the accession number of the entire gene and the protein used for this exercise.
(optional) What does it encode? Is the protein secreted?
Part 2: Annotation of an unknown sequence
This sequence is obtained from the NCBI database: Escherichia coli K12 subst. W3110 (ref: EF136884.1). No gaps or spaces in the sequence can exist when working with this sequence in one or another annotation tool. Paste this sequence into a Notebook or Word file and save it as a .txt file for use.
Learning goals:
- Verify the presence of a gene by identifying its key components.
- Identify critical components of an operon.
- Predict the function of a protein-based on its gene sequence.
- Describe the basic steps for annotating a genome.
This exercise will guide you through these first steps of manual genome annotation. You have been given an unknown sequence (part of a genome) to decipher the number and putative function of any potential genes in this sequence. Follow the steps below to complete the activity.
Obtain your nucleotide sequence. It is approximately 5500 nucleotides long. This is a single-stranded representative of a double-stranded DNA chromosome. Make sure you have the sequence in a .txt file. In addition, make sure there are no spaces or gaps between the nucleotides. This can result in inaccurate analysis by the gene-finding program we will use.
Here you have an open-end evaluation process!
One option may be based on the analysis of sequence similarity. It may only be possible because we work with the famous model organism with multiple genomes sequenced and deposited. Consequently, the search using the blast algorithm may be helpful. However, be prepared to spend some time locating your tiny 5500 nt-long fragments in the reference sequence you get by the blast. It would be tedious manual work. If you go this way, please retrieve all nt sequences for your detected proteins and align them to the target sequence (you may also wish to work on aa level, then do translation and consider all six reading frames). Always record accession numbers!
Another option is to use a computer program developed for this purpose. For example, you may use the program Artemis: Genome Browser and Annotation Tool (Rutherford et al., 2000 Sanger Institute). Artemis is a free genome browser and annotation tool that allows the visualization of sequence features, next-generation data, and the results of analyses within the sequence context and its six-frame translation.
Artemis is written in Java and is available for UNIX, Macintosh, and Windows systems. It can read EMBL and GENBANK database entries or sequences in FASTA, indexed FASTA, or raw format. Other sequence features can be in EMBL, GENBANK, or GFF format. This free program uses a mathematical algorithm to identify potential genes in the genome sequence. In addition, it will identify the translation start and stop sites for you.
Find Artemis: Genome Browser and Annotation Tool (locate yourself), click on the “Download” tab. You will be directed to another page where you can download the software onto your computer or launch it directly. To avoid downloading the program, click on the button that says “Launch Artemis.”
A small screen with the program title (Artemis) will open. Click on File and then on Open. Find and open your unknown sequence (The program automatically looks for sequence files. You will have to change the files it looks for by changing the search to “all files” instead of “sequence files.”) If successful, you should see your sequence in the program.
To briefly describe what is seen in Artemis, there are three rows containing black lines at the top of the screen, then two gray bars, followed by another three sets of rows containing the black lines. Each row represents one of the six frames for translation on the sequence. The top three represent the frames for reading the codons on the top strand of DNA, while the bottom three represent the frames for reading the codons on the bottom strand of DNA. The black lines represent stop codons within that reading frame. Use the right scroll bar to focus on the image and focus back out. At the bottom of the program is the information in more detail. The six reading frames are represented, and the amino acid code for all six translational frames is included. In addition, the sequence is located here as well. You can use the bottom scroll bar to move along the entire sequence. You can use the right scrollbar to focus on an area of focus on an area.
Identify putative genes in your unknown sequence:
Let us use the program to find putative genes in your sequence.
To activate the algorithm to identify putative genes, find the “create” tab. Then, scroll down and select “mark open reading frames…” (an open reading frame or ORF is another term for a putative gene).
Artemis will ask the minimum size for the open reading (how many amino acids long?). In order to prevent the identification of very small genes (that are likely not genes but are merely a start and stop codon in close proximity), we are going to search for open reading frames over 200 amino acids long. Type in 200.
Any identified ORFs or putative genes should be highlighted in blue. How many putative genes do you see?
Let us verify the start codon. As shown by the many black lines, the program does an excellent job identifying stop codons. First, however, it is essential to look at what start codon is being used. For example, some bacteria have a preference for certain stop codons. In this scenario, we will look for the traditional ATG start codon.
In the bottom (more magnified) window, scroll (using the bottom scroll bar) to the front of the first gene or double click on the first gene in the top window. This will highlight the gene sequence in the bottom window. Does it start with an ATG start codon? If not, you can “trim” the gene to our desired start codon.
If the gene does not start with ATG, highlight the gene (by clicking on it). Now locate the “edit” tab at the top of the program. Next, select the “trim selected feature.” Further, select “trim to met.”
Do this for your other genes.
How can we verify that these might be genes?
The Artemis program has found the start and stop sites for the translation of each gene. What other structures/sequences might you look for to help verify that these are genes? List any structures you might look for here:
You might have decided to look for the Shine Dalgarno and promoter regions. Here is a guide to examining the genes for these important gene features.
Find the Shine-Dalgarno region:
Where would the Shine Dalgarno be present on a gene? Go to this area of the gene in the bottom window. For example, look for the following Shine Dalgarno sequences: AGGA or AGCA.
- Should a Shine Dalgarno be present for each gene? Why or why not?
- Did you find one for each gene? What does this information provide?
Find the promoter region:
Where would the promoter be present on a gene? Go to this area of the gene in the bottom window.
- Although this genome may have different promoter consensus sequences, we will use a set of Escherichia coli consensus sequences.
(-35 sequence: TACACT; -10 sequence: TATGTT). Note that they are rich in T’s and A’s.
- Must you find a promoter for each gene? Why or why not?
- Did you find a promoter for each gene? If not, what does this suggest?
Are these genes?
Based on the evidence you have collected so far, reflect on whether you believe the identified genes are indeed genes. Then, provide evidence to support your claim.
What is the putative function of these genes?
Now that you have finished a basic analysis of your gene nucleotide sequence, it is time to examine the putative function of the encoded product of these genes. What do these proteins do for the cell? Will you trust your prediction, or will laboratory research still be required to verify the result?
Collect the protein sequence for your genes to conduct your comparison.
Because you have the gene sequence, you could use the codon tables to read the predicted amino acid sequence for this gene. However, in Artemis, the program will do the translation for you. Click on the first gene (so it is highlighted). On the menu, select the “View” tab and click on “amino acids of selection.” This will open a separate page with the primary amino acid sequence for the highlighted gene. Highlight and copy these nucleotides.
Compare your protein sequence with a database of proteins whose functions have been verified.
What is the protein name that it is similar to?
What organism does this protein come from (you can find this information at the top of the alignment)?
Reflect on what this information tells you? is it likely that your protein has a function similar to the aligned protein from the database? Please provide evidence to support your claim.
Function information
Use the protein information that you have received from your alignment to begin your research. What is the function of your protein(s)? One excellent database to help predict an E. coli protein’s function is EcoCyc. Curators of the database use the scientific literature based on experimental data to compile a metabolic and functional description of the Escherichia coli genome. Experimental data include protein structure data, enzymatic function, regulation of these gene products, and construction of metabolic pathways within the organism. Should your putative proteins match this database, much can be learned and predicted regarding its function.
Go to the EcoCyc webpage: Type in the name of each putative protein you have identified (or the four-letter gene name) into the box on the right. Then, click on a quick search. If a similar protein has been identified in the E. coli K-12 genome, these should be listed after the search under “proteins.” Next, click on the name of your protein.
What will the results tell you? First, you should see information regarding the regulation of this gene and information regarding the protein and its location and function. Research as much as you can regarding the putative proteins you have identified. Describe their putative functions here, based on the information you can find on the EcoCyc database.
Complete a functional analysis (BLAST and EcoCyc analysis) for all of your putative proteins. Please address: do these proteins have anything in common regarding their function? Might they be related?
Discuss any mechanisms of regulation that might exist for these proteins.
中文练习说明 Chinese version