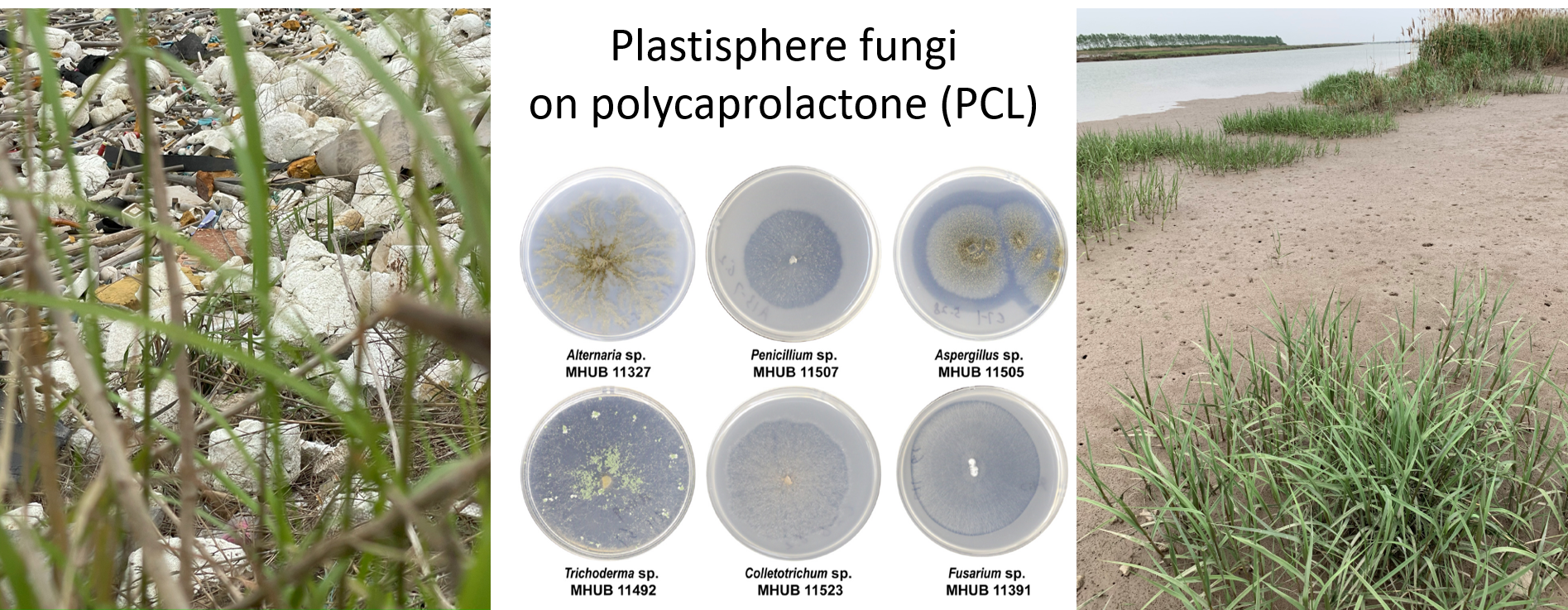
cf. Alternaria TUCIM11330
>11330ITS
GGTTAAGGTGACTGCGGGAGGGTCATTACACAAATATGAAGGCGGGCTGGAACCTCTCGGGGTTACAGCCTTGCTGAATTATTCACCCTTGTCTTTTGCGTACTTCTTGTTTCCTTGGTGGGTTCGCCCACCACTAGGACAAACATAAACCTTTTGTAATTGCAATCAGCGTCAGTAACAAATTAATAATTACAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCGAAATGCGATAAGTAGTGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCTTTGGTATTCCAAAGGGCATGCCTGTTCGAGCGTCATTTGTACCCTCAAGCTTTGCTTGGTGTTGGGCGTCTTGTCTCTAGCTTTGCTGGAGACTCGCCTTAAAGTAATTGGCAGCCGGCCTACTGGTTTCGGAGCGCAGCACAAGTCGCACTCTCTATCAGCAAAGGTCTAGCATCCATTAAGCCTTTTTTTCAACTTTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAAGCATATCAATAAGCGGGGAGAAAAC
>11330rpb2
GTGTACGGGCGATTGGTTGTGATCGGGGAAGGGAATGATACTGGCACAGATACCGAGAATCATAGCCGGATGAATCTCGCAATGAGTATAGGCATGGATGCGGGGGTCTGGTAGTGGCTTGAGGCGTCGGAGACGGTGCTCACCCTCGGTGGATCGCTCCGCCGCCGGCAAGCCCAATTTCATCTCTCGCCACTCCTCCAGATCCTCAGGGGAGAACGTTATCATGGCGGTCTCCTCCTCTTCAGCGTCAAGGTACTCCACAACACCGTCTTGAATGAGACCTCTCCAGCCGTAGGTAGCTGATTCGACCTCATCCTGGCTCCAACCCTGTCGTGTGCTTGTCTCTTGTTGCTCCTGCTTGAGCTTGTTACTGATCTCCTTGGTGAAGATGAGGTGGTTGCGGTTTGGCTTCCGAATGTCGTTCTCAACAACGAACAGTGGTCTCATGACACGGCCAGCATCTGTGAAGATCTTGAACTCTCGGTCACGAATGTCACGAATCAGACTCATCTCATAGGATAGAGTTCCGTTTCGTCGCAGCTCCTGCACAACTGTGACAAGTTGTTGAGCGTTGGAATGGACACCAACCCAGACACCGTTGACGAAGACCTTGGTGGCATCGGGGTTCTGGTTCTGATCATACTCTTCTAGAAGTTGCATGTTTCGTTGTGTCATGAAGTCGATGATGGGAGAAGCGTCACTACCGACACTAACATAGCACATCAAAGACAGGTTCTTAACCAGACCACAAGCCTGTCCTTCAGGAGTCTCGGCAGGGCAGACAAGACCCCAATGAGAGTTGTGGAGTTGTCGCGGCTTGGCCAGCTTTCCATCACGACCAACAGGGGTATTCGTTCGACGCAAATGGGACAGTGTGGAGGCCGTAAGTGTATCGTCACACC
>11330tef1
GATCTTTTCTTTACGCGGCTGTTGCGCCCACCCGGTGCTTTCTCTGAGCGCGTAGCCAAAGCCTGGCTTATCGCGATGAGGGGCATTTTTGGGTGGTGGGGATGTGCGAACTTTTACGCGCTAGCGCTAGTCCGCATGCGGCCTTCGCGAACTCCAACGCAATGACGCACATGTAATTTCCCCATTCTGGCCACAGCGAGCTAACAAGCCTCACAGGAAGCCGCCGAACTCGGTAAGGTTCCCCTTCAAGTAAA
>11330alt
TTGGCGTCGCTGGCTTGCCGCCGCTGCACCTCTCGAGTCTCGCCAGGACACCGCATCCTGCCCTGTCACCACCGAGGGTGACTACGTCTGGAAGATTTCCGAGTTCTACGGACGCAAGCCGGAGGGAACCTACTACAACAGCCTCGGCTTCAACATCAAGGCTACTAACGGAGGAACACTCGACTTCACCTGCTCTCACTCAGCCGACAAGCTTGAGGACCACACTTGGTACTCTTGCGGCGAGAACAGCTTCATGGACTTCTCTTTCGACAGCGACCGCAACGGTCTGCTCCTGAAGCAGAAGGTTAGCGACGAGTAAGTTACCCTTGTACCTTCGATTACTTCGCAGATTCAGATATACTAACATGTTTCCAGCATCACCTATGTCGCTACCGCCACTCTTCCCAACTACTGCCGCGCTGGCGGTAACGGCCCTAAGGACTTTGTCTGCCAGGGTGTTGCCGACGCCTAGC
>11330gapdh
GGCTGTCCTTCGATGCGTAGTTTCGCCTAACTCGTCGATACAATCTACCAGAGCTGACCGCATGCCACAGTATCGAGCACAACGACGTCGACATTGTCGCCGTAAACGACCCCTTCATCGAGCCCCACTACGCTGTAAGCTTCCCCAAGCACCCACACTACAGCCGCGGCCATCCAAGTTGCGAAAACAGTCCTTGCGATGCGCTAGAGCTCCTCTGTGGTCGCAGAATGCAGGCTAACACATTCAGGCCTACATGCTCAAGTATGACAGCACACACGGCCAGTTCAAGGGTGAGATCAAGGTTGACGGCAACAACCTGACCGTCAACGGCAAGACCATCCGTTTCCACATGGAGAAGGACCCCGCCAACATCCCATGGAGCGAGACCGGCGCTTACTACGTCGTCGAGTCCACCGGTGTCTTCACCACCACCGAGAAGGCCAAGGCTCACTTGAAGGGTGGAGCCAAGAAGGTCGTCATTTCTGCTCCCTCTGCTGACGCCCCCATGTTCGTTATGGGTGTCAACCACGAGACTTACAAGTCTGACATCGAGGTTCTCTCCAACGCCTCTTGCACAACCACTTTTGCTTTGGCGGA
cf. Alternaria TUCIM11332
>11332ITS
CGACGGAGGGATCTTACACAAATATGAAGGCGGGCTGGAACCTCTCGGGGTTACAGCCTTGCTGAATTATTCACCCTTGTCTTTTGCGTACTTCTTGTTTCCTTGGTGGGTTCGCCCACCACTAGGACAAACATAAACCTTTTGTAATTGCAATCAGCGTCAGTAACAAATTAATAATTACAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCGAAATGCGATAAGTAGTGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCTTTGGTATTCCAAAGGGCATGCCTGTTCGAGCGTCATTTGTACCCTCAAGCTTTGCTTGGTGTTGGGCGTCTTGTCTCTAGCTTTGCTGGAGACTCGCCTTAAAGTAATTGGCAGCCGGCCTACTGGTTTCGGAGCGCAGCACAAGTCGCACTCTCTATCAGCAAAGGTCTAGCATCCATTAAGCCTTTTTTTCAACTTTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAAGCATATCAATAAGCGGGAAAAAAA
>11332rpb2
GTTAGGCGGGTGTCTGCAGGTGTTGACCGATACACTTACGCCTCCACACTGTCACATTTGCGTCGAACGAATACCCCTGTTGGTCGTGATGGAAAGCTGGCCAAGCCGCGACAACTCCACAACTCTCATTGGGGTCTTGTCTGCCCTGCCGAGACTCCTGAAGGACAGGCTTGTGGTCTGGTTAAGAACCTGTCTTTGATGTGCTATGTTAGTGTCGGTAGTGACGCTTCTCCCATCATCGACTTCATGACACAACGAAACATGCAACTCCTAGAAGAGTATGATCAGAACCAGAACCCCGATGCCACCAAGGTCTTTGTCAACGGTGTCTGGGTTGGTGTCCATTCCAACGCTCAACAACTTGTCACAGTTGTGCAGGAGCTGCGACGAAACGGAACTCTATCCTATGAGATGAGTCTGATTCGTGACATTCGTGACCGAGAGTTCAAGATCTTCACAGATGCTGGCCGTGTCATGAGACCACTGTTCGTTGTTGAGAACGACATTCGGAAGCCAAACCGCAACCACCTCATCTTCACCAAGGAGATCAGTAACAAGCTCAAGCAGGAACAACAAGAGACAAGCACACGACAGGGTTGGAGCCAGGATGAGGTCGAATCAGCTACCTACGGCTGGAGAGGTCTTATTCAAGACGGTGTTGTGGAGTACCTAGACGCTGAAGAGGAGGAGACCGCCATGATAACGTTCTCCCCTGAGGATCTGGAGGAGTGGCGAGAGATGAAATTGGGTTTGCCGGCGGCGGAGCGATCCACCGAGGGTGAGCACCGTCTCCGACGCCTCAAGCCACTACCAGACCCCCGCATCCATGCCTATACTCATTGCGAGATTCATCCGGCTATGATTCTCGGTATCTGTGCCAGTATCATTCCCTTCCCCGATCACAACCAATCGCCCCGAACGGGCA
>11332tef1
AACTAACTCTTTCGCGGCCTGTTGCGCCCACCCGGTGCTTTCTCTGAGCGCGTAGCCAAAGCCTGGCTTATCGCGATGAGGGGCATTTTTGGGTGGTGGGGATGTGCGAACTTTTACGCGCTAGCGCTAGTCCGCATGCGGCCTTCGCGAACTCCAACGCAATGACGCACATGTAATTTCCCCATTCTGGCCACAGCGAGCTAACAAGCCTCACAGGAAGCCGCCGAACTCGGTAAGGCCCCCTTTC
>11332alt
GTCGGCAACACCCTGGCAGACAAAGTCCTTAGGGCCGTTACCGCCAGCGCGGCAGTAGTTGGGAAGAGTGGCGGTAGCGACATAGGTGATGCTGGAAACATGTTAGTATATCTGAATCTGCGAAGTAATCGAAGGTACAAGGGTAACTTACTCGTCGCTAACCTTCTGCTTCAGGAGCAGACCGTTGCGGTCGCTGTCGAAAGAGAAGTCCATGAAGCTGTTCTCGCCGCAAGAGTACCAAGTGTGGTCCTCAAGCTTGTCGGCTGAGTGAGAGCAGGTGAAGTCGAGTGTTCCTCCGTTGGTAGCCTTGATGTTGAAGCCGAGGCTGTTGTAGTAGGTTCCCTCCGGCTTGCGTCCGTAGAACTCGGAAATCTTCCAGACGTAGTCACCCTCGGTGGTGACAGGGCAGGATGCGGTGTCCTGGCGAGACTCGAGAGGTGCAGCGGCGGCAAGGCCAGCGGCGGCGAAGAGAGAGGC
>11332gapdh
TGGGTGGAGACTCGATGTAGACTTGTAGTCTCGTGGTTGACACCCATAACGAACATGGGGGCGTCAGCAGAGGGAGCAGAAATGACGACCTTCTTGGCTCCACCCTTCAAGTGAGCCTTGGCCTTCTCGGTGGTGGTGAAGACACCGGTGGACTCGACGACGTAGTAAGCGCCGGTCTCGCTCCATGGGATGTTGGCGGGGTCCTTCTCCATGTGGAAACGGATGGTCTTGCCGTTGACGGTCAGGTTGTTGCCGTCAACCTTGATCTCACCCTTGAACTGGCCGTGTGTGCTGTCATACTTGAGCATATAGGCCTGAATGTGTTAGCCTGCATTCTGCGACCACAGAGGAGCTCTAGCGCATCGCAAGGACTGTTTTCGCAACTTGGATGGCCGCGGCTGTAGTGTGGGTGCTTGGGGAAGCTTACAGCGTAGTGGGGCTCGATGAAAGGGTCGTTTACGGCGACAATGTCGACGTCGTTGTGCTCGATACTGTGGCATGCGGTCAGCTCTGGTAGATTGTATCGACGAGTTAGGCGAAACTTACGCATTGCGGAAGACGATACGGCCAATGCGACGAAAACCCCGTTGAAAAACC
cf. Alternaria TUCIM11326
>11326ITS
CCTTTAGGGTACTGCGGAGGGTCATTACACAAATATGAAGGCGGGCTGGAACCTCTCGGGGTTACAGCCTTGCTGAATTATTCACCCTTGTCTTTTGCGTACTTCTTGTTTCCTTGGTGGGTTCGCCCACCACTAGGACAAACATAAACCTTTTGTAATTGCAATCAGCGTCAGTAACAAATTAATAATTACAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCGAAATGCGATAAGTAGTGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCTTTGGTATTCCAAAGGGCATGCCTGTTCGAGCGTCATTTGTACCCTCAAGCTTTGCTTGGTGTTGGGCGTCTTGTCTCTAGCTTTGCTGGAGACTCGCCTTAAAGTAATTGGCAGCCGGCCTACTGGTTTCGGAGCGCAGCACAAGTCGCACTCTCTATCAGCAAAGGTCTAGCATCCATTAAGCCTTTTTTTCAACTTTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAAGCATATCAATAAGCGGGAAAAAAAA
>11326rpb2
GCCGTGGCGGTGTCTCATGTGTGACGATACACTTACGCCTCCACACTGTCACATTTGCGTCGAACGAATACCCCTGTTGGTCGTGATGGAAAGCTGGCCAAGCCGCGACAACTCCACAACTCTCATTGGGGTCTTGTCTGCCCTGCCGAGACTCCTGAAGGACAGGCTTGTGGTCTGGTTAAGAACCTGTCTTTGATGTGCTATGTTAGTGTCGGTAGTGACGCTTCTCCCATCATCGACTTCATGACACAACGAAACATGCAACTCCTAGAAGAGTATGATCAGAACCAGAACCCCGATGCCACCAAGGTCTTTGTCAACGGTGTCTGGGTTGGTGTCCATTCCAACGCTCAACAACTTGTCACAGTTGTGCAGGAGCTGCGACGAAACGGAACTCTATCCTATGAGATGAGTCTGATTCGTGACATTCGTGACCGAGAGTTCAAGATCTTCACAGATGCTGGCCGTGTCATGAGACCACTGTTCGTTGTTGAGAACGACATTCGGAAGCCAAACCGCAACCACCTCATCTTCACCAAGGAGATCAGTAACAAGCTCAAGCAGGAACAACAAGAGACAAGCACACGACAGGGTTGGAGCCAGGATGAGGTCGAATCAGCTACCTACGGCTGGAGAGGTCTTATTCAAGACGGTGTTGTGGAGTACCTAGACGCTGAAGAGGAGGAGACCGCCATGATAACGTTCTCCCCTGAGGATCTGGAGGAGTGGCGAGAGATGAAATTGGGTTTGCCGGCGGCGGAGCGATCCACCGAGGGTGAGCACCGTCTCCGACGCCTCAAGCCACTACCAGACCCCCGCATCCATGCCTATACTCATTGCGAGATTCATCCGGCTATGATTCTCGGTATCTGTGCCAGTATCATTCCCTTCCCCGATCACAACCAATCGCCCCGTAACACCTGAGTCCC
>11326tef1
GGGTAACTTTTCTTACGCGGCCTGTTGCGCCCACCCGGTGCTTTCTCTGAGCGCGTAGCCAAAGCCTGGCTTATCGCGATGAGGGGCATTTTTGGGTGGTGGGGATGTGCGAACTTTTACGCGCTAGCGCTAGTCCGCATGCGGCCTTCGCGAACTCCAACGCAATGACGCACATGTAATTTCCCCATTCTGGCCACAGCGAGCTAACAAGCCTCACAGGAAGCCGCCGAACTCGGTAAGGTTCCCCTTCAAGTAAA
>11326alt
CTGCGGTCGCTGGCTTGCCGCTGCTGCTCCTCTCGAGTCTCGCCAGGACACCGCATCCTGCCCTGTCACCACTGAGGGTGACTACGTCTGGAAGATCTCCGAATTCTACGGACGCAAGCCGGAAGGAACCTACTACAACAGCCTCGGCTTCAACATCAAGGCCACCAACGGAGGAACCCTCGACTTCACCTGCTCTGCTCAGGCCGATAAGCTTGAGGACCACAAGTGGTACTCCTGTGGCGAGAACAGCTTCATGGACTTCTCTTTCGACAGCGACCGCAGCGGTCTGCTCCTGAAGCAGAAGGTTAGCGACGAGTAAGTTACCCTTGTACCTTCGATTACTTCGCAGATTCAGATATACTAACATATCCCCAGCATCACCTATGTCGCTACCGCCACTCTTCCCAACTACTGCCGCGCTGGCGGTAACGGCCCTAAGGACTTTGTCTGCCAGGGTGTTGCCGACGCCA
>11326gapdh
TGACGTTGAGACTCGATGTAGACTTGTAGTCTCGTGGTTGACACCCATAACGAACATGGGGGCGTCAGCAGAGGGAGCAGAAATGACGACCTTCTTGGCTCCACCCTTCAAGTGAGCCTTGGCCTTCTCGGTGGTGGTGAAGACACCGGTGGACTCGACGACGTAGTAAGCGCCGGTCTCGCTCCATGGGATGTTGGCGGGGTCCTTCTCCATGTGGAAACGGATGGTCTTGCCGTTGACGGTCAGGTTGTTGCCGTCAACCTTGATCTCACCCTTGAACTGGCCGTGTGTGCTGTCATACTTGAGCATGTAGGCCTGAATGTGTTAGCCTGCATTCTGCGACCACAGAGGAGCTCTAGCGCATCGCAAGGACTGTTTTCGCAACTTGGATGGCCGCGGCTGTAGTGTGGGTGCTTGGGGAAGCTTACAGCGTAGTGGGGCTCGATGAAGGGGTCGTTTACGGCGACAATGTCGACGTCGTTGTGCTCGATACTGTGGCATGCGGTCAGCTCTGGTAGATTGTATCGACGAGTTAGGCGAAACTTACGCATTGCGGAAGACGATACGGCCATGCCGAAAAAAAACCCTTTTTTGGGGGG
cf. Alternaria TUCIM11321
>11321ITS
GCCGAGGGGACTGGCGGAGGGTCATTACACAAATATGAAGGCGGGCTGGAACCTCTCGGGGTTACAGCCTTGCTGAATTATTCACCCTTGTCTTTTGCGTACTTCTTGTTTCCTTGGTGGGTTCGCCCACCACTAGGACAAACATAAACCTTTTGTAATTGCAATCAGCGTCAGTAACAAATTAATAATTACAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCGAAATGCGATAAGTAGTGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCTTTGGTATTCCAAAGGGCATGCCTGTTCGAGCGTCATTTGTACCCTCAAGCTTTGCTTGGTGTTGGGCGTCTTGTCTCTAGCTTTGCTGGAGACTCGCCTTAAAGTAATTGGCAGCCGGCCTACTGGTTTCGGAGCGCAGCACAAGTCGCACTCTCTATCAGCAAAGGTCTAGCATCCATTAAGCCTTTTTTTCAACTTTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAAGCATATCAATAAGCGGGGAAGAAAA
>11321rpb2
GCCGGGAGGCGATGTCTCCAGGTGTGACCGATACACTTACGCCTCCACACTGTCCCATTTGCGTCGAACGAATACCCCTGTTGGTCGTGATGGAAAGCTGGCCAAGCCGCGACAACTCCACAACTCTCATTGGGGTCTTGTCTGCCCTGCCGAGACTCCTGAAGGACAGGCTTGTGGTCTGGTTAAGAACCTGTCTTTGATGTGCTATGTTAGTGTCGGTAGTGACGCTTCTCCCATCATCGACTTCATGACACAACGAAACATGCAACTTCTAGAAGAGTATGATCAGAACCAGAACCCCGATGCCACCAAGGTCTTCGTCAACGGTGTCTGGGTTGGTGTCCATTCCAACGCTCAACAACTTGTCACAGTTGTGCAGGAGCTGCGACGAAACGGAACTCTATCCTATGAGATGAGTCTGATTCGTGACATTCGTGACCGAGAGTTCAAGATCTTCACAGATGCTGGCCGTGTCATGAGACCACTGTTCGTTGTTGAGAACGACATTCGGAAGCCAAACCGCAACCACCTCATCTTCACCAAGGAGATCAGTAACAAGCTCAAGCAGGAGCAACAAGAGACAAGCACACGACAGGGTTGGAGCCAGGATGAGGTCGAATCAGCTACCTACGGCTGGAGAGGTCTCATTCAAGACGGTGTTGTGGAGTACCTTGACGCTGAAGAGGAGGAGACCGCCATGATAACGTTCTCCCCTGAGGATCTGGAGGAGTGGCGAGAGATGAAATTGGGCTTGCCGGCGGCGGAGCGATCCACCGAGGGTGAGCACCGTCTCCGACGCCTCAAGCCACTACCAGACCCCCGCATCCATGCCTATACTCATTGCGAGATTCATCCGGCTATGATTCTCGGTATCTGTGCCAGTATCATTCCCTTCCCCGATCACAACCAATCGCCCCGTACACATGCCTCC
>11321tef1
GGGTATCCTTTTCTTTACGCGGCCTGTTGCGCCCACCCGGTGCTTTCTCTGAGCGCGTAGCCAAAGCCTGGCTTATCGCGATGAGGGGCATTTTTGGGTGGTGGGGATGTGCGAACTTTTACGCGCTAGCGCTAGTCCGCATGCGGCCTTCGCGAACTCCAACGCAATGACGCACATGTAATTTCCCCATTCTGGCCACAGCGAGCTAACAAGCCTCACAGGAAGCCGCCGAACTCGGTAAGGGTCCCCTTTCAAGTA
>11321alt
CTTGGGCCGCTGGCTTGCCGCTGCTGCTCCTCTCGAGTCTCGCCAGGACACCGCATCCTGCCCTGTCACCACTGAGGGTGACTACGTCTGGAAGATCTCCGAATTCTACGGACGCAAGCCGGAAGGAACCTACTACAACAGCCTCGGCTTCAACATCAAGGCCACCAACGGAGGAACCCTCGACTTCACCTGCTCTGCTCAGGCCGATAAGCTTGAGGACCACAAGTGGTACTCCTGTGGCGAGAACAGCTTCATGGACTTCTCTTTCGACAGCGACCGCAGCGGTCTGCTCCTGAAGCAGAAGGTTAGCGACGAGTAAGTTACCCTTGTACCTTCGATTACTTCGCAGATTCAGATATACTAACATATTCCCAGCATCACCTATGTCGCTACCGCCACTCTTCCCAACTACTGCCGCGCTGGCGGTAACGGCCCTAAGGACTTTGTCTGCCAGGGTGTTGCCGACGCCTACTC
>11321gapdh
CGTTGGAGACTCGATGTAGACTTGTAAGTCTCGTGGTTGACACCCATAACGAACATGGGGGCGTCAGCAGAGGGAGCAGAAATGACGACCTTCTTGGCTCCACCCTTCAAGTGAGCCTTGGCCTTCTCGGTGGTGGTGAAGACACCGGTGGACTCGACGACGTAGTAAGCGCCGGTCTCGCTCCATGGGATGTTGGCGGGGTCCTTCTCCATGTGGAAACGGATGGTCTTGCCGTTGACGGTCAGGTTGTTGCCGTCAACCTTGATCTCACCCTTGAACTGGCCGTGTGTGCTGTCATACTTGAGCATGTAGGCCTGAATGTGTTAGCCTGCATTCTGCGACCACAGAGGAGCTCTAGCGCATCGCAAGGACTGTTTTCGCAACTTGGATGGCCGCGGCTGTAGTGTGGGTGCTTGGGGAAGCTTACAGCGTAGTGGGGCTCGATGAAGGGGTCGTTTACGGCGACAATGTCGACGTCGTTGTGCTCGATACTGTGGCATGCGGTCAGCTCTGGTAGATTGTATCGACGAGTTAGGCGAAACTTACGCATTGCGGAAGACGATACGGCCATGCCGAAAAAAAACCCCCCTGGGGGG
cf. Alternaria TUCIM11323
>11323ITS
GCCTAAGGTGACTGCGGAGGGTCATTACACAAATATGAAGGCGGGCTGGAACCTCTCGGGGTTACAGCCTTGCTGAATTATTCACCCTTGTCTTTTGCGTACTTCTTGTTTCCTTGGTGGGTTCGCCCACCACTAGGACAAACATAAACCTTTTGTAATTGCAATCAGCGTCAGTAACAAATTAATAATTACAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCGAAATGCGATAAGTAGTGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCTTTGGTATTCCAAAGGGCATGCCTGTTCGAGCGTCATTTGTACCCTCAAGCTTTGCTTGGTGTTGGGCGTCTTGTCTCTAGCTTTGCTGGAGACTCGCCTTAAAGTAATTGGCAGCCGGCCTACTGGTTTCGGAGCGCAGCACAAGTCGCACTCTCTATCAGCAAAGGTCTAGCATCCATTAAGCCTTTTTTTCAACTTTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAAGCATATCAATAAGCGAGGAAGAA
>11323rpb2
GGCTGGCGGTTGTGTCTCAGTGTGACCGATACACTTACGCCTCCACACTGTCCCATTTGCGTCGAACGAATACCCCTGTTGGTCGTGATGGAAAGCTGGCCAAGCCGCGACAACTCCACAACTCTCATTGGGGTCTTGTCTGCCCTGCCGAGACTCCTGAAGGACAGGCTTGTGGTCTGGTTAAGAACCTGTCTTTGATGTGCTATGTTAGTGTCGGTAGTGACGCTTCTCCCATCATCGACTTCATGACACAACGAAACATGCAACTTCTAGAAGAGTATGATCAGAACCAGAACCCCGATGCCACCAAGGTCTTCGTCAACGGTGTCTGGGTTGGTGTCCATTCCAACGCTCAACAACTTGTCACAGTTGTGCAGGAGCTGCGACGAAACGGAACTCTATCCTATGAGATGAGTCTGATTCGTGACATTCGTGACCGAGAGTTCAAGATCTTCACAGATGCTGGCCGTGTCATGAGACCACTGTTCGTTGTTGAGAACGACATTCGGAAGCCAAACCGCAACCACCTCATCTTCACCAAGGAGATCAGTAACAAGCTCAAGCAGGAGCAACAAGAGACAAGCACACGACAGGGTTGGAGCCAGGATGAGGTCGAATCAGCTACCTACGGCTGGAGAGGTCTCATTCAAGACGGTGTTGTGGAGTACCTTGACGCTGAAGAGGAGGAGACCGCCATGATAACGTTCTCCCCTGAGGATCTGGAGGAGTGGCGAGAGATGAAATTGGGCTTGCCGGCGGCGGAGCGATCCACCGAGGGTGAGCACCGTCTCCGACGCCTCAAGCCACTACCAGACCCCCGCATCCATGCCTATACTCATTGCGAGATTCATCCGGCTATGATTCTCGGTATCTGTGCCAGTATCATTCCCTTCCCCGATCACAACCAATCGCCCCGTACACCTGACTTC
>11323tef1
GGGGTCGAATCTTACGCGGCCTGTTGCGCCCACCCGGTGCTTTCTCTGAGCGCGTAGCCAAAGCCTGGCTTATCGCGATGAGGGGCATTTTTGGGTGGTGGGGATGTGCGAACTTTTACGCGCTAGCGCTAGTCCGCATGCGGCCTTCGCGAACTCCAACGCAATGACGCACATGTAATTTCCCCATTCTGGCCACAGCGAGCTAACAAGCCTCACAGGAAGCCGCCGAACTCGGTAAGGTCCCCTTTCAAGTAAAC
>11323alt
TTGCGACGCTGGACTTGCCGCCGCTGCACCTCTCGAGTCTCGCCAGGACACCGCATCCTGCCCTGTCACCACCGAGGGTGACTACGTCTGGAAGATTTCCGAGTTCTACGGACGCAAGCCGGAGGGAACCTACTACAACAGCCTCGGCTTCAACATCAAGGCTACCAACGGAGGAACACTCGACTTCACCTGCTCTCACTCAGCCGACAAGCTTGAGGACCACACTTGGTACTCTTGCGGCGAGAACAGCTTCATGGACTTCTCTTTCGACAGCGACCGCAACGGTCTGCTCCTGAAGCAGAAGGTTAGCGACGAGTAAGTTACCCTTGTACCTTCGATTACTTCGCAGATTCAGATATACTAACATGTTTCCAGCATCACCTATGTCGCTACCGCCACTCTTCCCAACTACTGCCGCGCTGGCGGTAACGGCCCTAAGGACTTTGTCTGCCAGGGTGTTGCCGACGCCTAA
>11323gapdh
GGACGTGGGGACTCGATGTAGACTTGTAGTCTCGTGGTTGACACCCATAACGAACATGGGGGCGTCAGCAGAGGGAGCAGAAATGACGACCTTCTTGGCTCCACCCTTCAAGTGAGCCTTGGCCTTCTCGGTGGTGGTGAAGACACCGGTGGACTCGACGACGTAGTAAGCGCCGGTCTCGCTCCATGGGATGTTGGCGGGGTCCTTCTCCATGTGGAAACGGATGGTCTTGCCGTTGACGGTCAGGTTGTTGCCGTCAACCTTGATCTCACCCTTGAACTGGCCGTGTGTGCTGTCATACTTGAGCATATAGGCCTGAATGTGTTAGCCTGCATTCTGCGACCACAGAGGAGCTCTAGCGCATCGCAAGGACTGTTTTCGCAACTTGGATGGCCGCGGCTGTAGTGTGGGTGCTTGGGGAAGCTTACAGCGTAGTGGGGCTCGATGAAAGGGTCGTTTACGGCGACAATGTCGACGTCGTTGTGCTCGATACTGTGGCATGCGGTCAGCTCTGGTAGATTGTATCGACGAGTTAGGCGAAACTTACGCATTGCGGAAGACGATACGGCCATGGACGAAAAAAACCTTTTTTGGGGGG
cf. Alternaria TUCIM11325
>11325ITS
GCTTAGGGTACTGCGGAGGGTCATTACACAAATATGAAGGCGGGCTGGAACCTCTCGGGGTTACAGCCTTGCTGAATTATTCACCCTTGTCTTTTGCGTACTTCTTGTTTCCTTGGTGGGTTCGCCCACCACTAGGACAAACATAAACCTTTTGTAATTGCAATCAGCGTCAGTAACAAATTAATAATTACAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCGAAATGCGATAAGTAGTGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCTTTGGTATTCCAAAGGGCATGCCTGTTCGAGCGTCATTTGTACCCTCAAGCTTTGCTTGGTGTTGGGCGTCTTGTCTCTAGCTTTGCTGGAGACTCGCCTTAAAGTAATTGGCAGCCGGCCTACTGGTTTCGGAGCGCAGCACAAGTCGCACTCTCTATCAGCAAAGGTCTAGCATCCATTAAGCCTTTTTTTCAACTTTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAAGCATATCAATAAGCGATAAAAAAAAA
>11325rpb2
GCCGTCGCTGTGTCTCAGGTGTGACGATACACTTACGCCTCCACACTGTCCCATTTGCGTCGAACGAATACCCCTGTTGGTCGTGATGGAAAGCTGGCCAAGCCGCGACAACTCCACAACTCTCATTGGGGTCTTGTCTGCCCTGCCGAGACTCCTGAAGGACAGGCTTGTGGTCTGGTTAAGAACCTGTCTTTGATGTGCTATGTTAGTGTCGGTAGTGACGCTTCTCCCATCATCGACTTCATGACACAACGAAACATGCAACTTCTAGAAGAGTATGATCAGAACCAGAACCCCGATGCCACCAAGGTCTTCGTCAACGGTGTCTGGGTTGGTGTCCATTCCAACGCTCAACAACTTGTCACAGTTGTGCAGGAGCTGCGACGAAACGGAACTCTATCCTATGAGATGAGTCTGATTCGTGACATTCGTGACCGAGAGTTCAAGATCTTCACAGATGCTGGCCGTGTCATGAGACCACTGTTCGTTGTTGAGAACGACATTCGGAAGCCAAACCGCAACCACCTCATCTTCACCAAGGAGATCAGTAACAAGCTCAAGCAGGAGCAACAAGAGACAAGCACACGACAGGGTTGGAGCCAGGATGAGGTCGAATCAGCTACCTACGGCTGGAGAGGTCTCATTCAAGACGGTGTTGTGGAGTACCTTGACGCTGAAGAGGAGGAGACCGCCATGATAACGTTCTCCCCTGAGGATCTGGAGGAGTGGCGAGAGATGAAATTGGGCTTGCCGGCGGCGGAGCGATCCACCGAGGGTGAGCACCGTCTCCGACGCCTCAAGCCACTACCAGACCCCCGCATCCATGCCTATACTCATTGCGAGATTCATCCGGCTATGATTCTCGGTATCTGTGCCAGTATCATTCCCTTCCCCGATCACAACCAATCGCCCCGTACAGAGATTC
>11325tef1
GGGTATCTTTTCTTACGCGGCCTGTTGCGCCCACCCGGTGCTTTCTCTGAGCGCGTAGCCAAAGCCTGGCTTATCGCGATGAGGGGCATTTTTGGGTGGTGGGGATGTGCGAACTTTTACGCGCTAGCGCTAGTCCGCATGCGGCCTTCGCGAACTCCAACGCAATGACGCACATGTAATTTCCCCATTCTGGCCACAGCGAGCTAACAAGCCTCACAGGAAGCCGCCGAACTCGGTAAGGTTCCCCTTCAAGTAAA
>11325alt
TTTGCGTCGCTGGCTTGCCGCCGCTGCACCTCTCGAGTCTCGCCAGGACACCGCATCCTGCCCTGTCACCACCGAGGGTGACTACGTCTGGAAGATTTCCGAGTTCTACGGACGCAAGCCGGAGGGAACCTACTACAACAGCCTCGGCTTCAACATCAAGGCTACCAACGGAGGAACACTCGACTTCACCTGCTCTCACTCAGCCGACAAGCTTGAGGACCACACTTGGTACTCTTGCGGCGAGAACAGCTTCATGGACTTCTCTTTCGACAGCGACCGCAACGGTCTGCTCCTGAAGCAGAAGGTTAGCGACGAGTAAGTTACCCTTGTACCTTCGATTACTTCGCAGATTCAGATATACTAACATGTTTCCAGCATCACCTATGTCGCTACCGCCACTCTTCCCAACTACTGCCGCGCTGGCGGTAACGGCCCTAAGGACTTTGTCTGCCAGGGTGTTGCCGACGCCTA
>11325gapdh
GGACGGGGAGACTCGATGTAGACTTGTAAGTCTCGTGGTTGACACCCATAACGAACATGGGGGCGTCAGCAGAGGGAGCAGAAATGACGACCTTCTTGGCTCCACCCTTCAAGTGAGCCTTGGCCTTCTCGGTGGTGGTGAAGACACCGGTGGACTCGACGACGTAGTAAGCGCCGGTCTCGCTCCATGGGATGTTGGCGGGGTCCTTCTCCATGTGGAAACGGATGGTCTTGCCGTTGACGGTCAGGTTGTTGCCGTCAACCTTGATCTCACCCTTGAACTGGCCGTGTGTGCTGTCATACTTGAGCATGTAGGCCTGAATGTGTTAGCCTGCATTCTGCGACCACAGAGGAGCTCTAGCGCATCGCAAGGACTGTTTTCGCAACTTGGATGGCCGCGGCTGTAGTGTGGGTGCTTGGGGAAGCTTACAGCGTAGTGGGGCTCGATGAAGGGGTCGTTTACGGCGACAATGTCGACGTCGTTGTGCTCGATACTGTGGCATGCGGTCAGCTCTGGTAGATTGTATCGACGAAGTTAGGCGAAACTTACGGCATTGCCGGAAGACGATACGGCCAATGCACGAAAAAACCCCCCTTTTG
cf. Apiospora TUCIM11524
>11524ITS
GGGCGTCTGGGGGAGAAGCGGAGGACATTACAGAGTTATACAACTCCCACACCATTTGCCAACTTTACTCAGTTATGCCTCGGCGTAAGCTCCGTACGGGGCTGCCGGGTTGCGCTGCGGGCGACAGCTACCCTGTAGCTTACCCTGTAGCGCTACCCTGTAGCGTTACCCTGCGGCGGCCCGCCGGTGGAAACGAAACTCTTGTTTTATTGTATCTTCTGAGCGTCTTATTTTAATAAGTTAAAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCGAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCATCAGTATTCTGGTGGGCATGCCTGTTCGAGCGTCATTTCAACCCTTAAGCCTAGCTTAGTGTTGGGAATCGACCGTAGGGTCGTTCCTTAAAGACAGTGGCGGAGCGGCAGTGGTCCTCTGAGCGTAGTAAATTTATTTCTCGCTTTTGTCAGGCCCTGTCCTCCCGCCATAAAACCCCCAATTTTTTAGTGGTTGACCTCGGATCAGGTAGGAATACCCGCTGAACTTAAGCATATCAATAAGCGGAGGAAAAAAAACACCCAACCAA
>11524tef1
GGCATGATACTGATACTCGCAGGCTGACTGCGCTATTCTCATCATTGCCGCTGGTACTGGTGAGTTCGAGGCTGGTATCTCCAAGGATGGCCAGACTCGTGAGCACGCTCTGCTCGCTTTCACCCTCGGTGTCAAGCAGCTCATCGTCGCCATCAACAAGATGGACACCACCAAGTGGTCTGAGGCCCGTTACAAGGAGATCATCAAGGAGACTTCCTCCTTCATCAAGAAGGTTGGCTACAACCCCAAGGAGGTCGCTTTCGTCCCCATCTCCGGTTTCCACGGCGACAACATGCTGGAGGAGTCCACCAACATGCCCTGGTACAAGGGTTGGGAGAAGGAGACCAAGGCTGGCGGCAAGAAGACCGGCAAGACCCTGTTCCAGGCCATCGATGCCATCAACCCTCCCCAGCGTCCCACCGACAAGCCCCTCCGTCTTCCCCTTCAGGATGTCTACAAGATCGGGATTTG
>11524tub
CGGGGATGAGACGGATTGACCGACACGAAGTTGTCGGGGCGGAAAAGCTGGCCGAAAGGACCGGCACGGACGGCGTCCATAGTACCAGGCTCCAGATCGACGAGGACGGCACGAGGAACGTACTTGTTGCCGGAAGCCTCGTTGAAGTAAACGCTCATGCGCTCCAGCTGGAGCTCAGAGGTACCGTTGTAGCTATCGAAGGGATCTGTCAGCAGGAAGCAAGCAAGGTGGCGTTGTACGGGCAAGACTTACACTCCATTGCTGTCGAGGCCGTGCTCGCTGGAAATCTGTTGCCTGTAAGGGGACACTAGTCAGTATCCTGTACCCCCCAAATTGATCGACGTAGATAGGCCGTCTCTGCCAGCTTGAATTTGTATCGCATTGAACCAGAACGAATTTGGGTCGATCGACGGGCACGAAGAAGGCGAAGTAGGTACGCACCAGAAAGCAGCCCATTTTT
cf. Aspergillus TUCIM11504
>11504ITS
ACGACTGCCGAGTGACTGCGGAGGACATTACCGAGTGTAGGGTTCCTAGCGAGCCCAACCTCCCACCCGTGTTTACTGTACCTTAGTTGCTTCGGCGGGCCCGCCATTCATGGCCGCCGGGGGCTCTCAGCCCCGGGCCCGCGCCCGCCGGAGACACCACGAACTCTGTCTGATCTAGTGAAGTCTGAGTTGATTGTATCGCAATCAGTTAAAACTTTCAACAATGGATCTCTTGGTTCCGGCATCGATGAAGAACGCAGCGAAATGCGATAACTAGTGTGAATTGCAGAATTCCGTGAATCATCGAGTCTTTGAACGCACATTGCGCCCCCTGGTATTCCGGGGGGCATGCCTGTCCGAGCGTCATTGCTGCCCATCAAGCACGGCTTGTGTGTTGGGTCGTCGTCCCCTCTCCGGGGGGGACGGGCCCCAAAGGCAGCGGCGGCACCGCGTCCGATCCTCGAGCGTATGGGGCTTTGTCACCCGCTCTGTAGGCCCGGCCGGCGCTTGCCGAACGCAAATCAATCTTTTCCAGGTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAAGCATATCAATAAGCGGAGGAAAAAAAAACCCAACCCAAATTC
>11504rpb2
AAGCTTAGGCTGCGTGTCGCAGTGCTCAGTCGTTATACCTACGCTTCTACTTTGTCACATCTTCGCCGAACCAATACGCCTATTGGTCGTGATGGTAAAATTGCTAAACCCCGCCAGCTTCACAATACTCATTGGGGGTTGGTTTGTCCAGCAGAGACTCCTGAAGGTCAGGCCTGTGGTCTGGTCAAGAACTTGGCCCTCATGTGTTATGTCACTGTTGGTACACCCAGCGAACCTATCATTGACTTCATGATTCAGCGTAACATGGAAGTTCTTGAGGAGTTTGAGCCTCAGGTCACTCCCAACGCAACGAAGGTCTTCGTCAATGGAGTGTGGGTCGGTATCCATAGAGATCCCGCCCATCTTGTCAATACGATGCAGTCACTGCGGCGACGAAATATGATCTCCCACGAGGTCAGTTTGATTCGTGATATCCGTGAGCGGGAGTTCAAGATCTTCACCGATGCTGGACGTGTTTGCAGGCCGTTGTTCGTCATTGACAATGACCCGAAGAGCGAAAATTGCGGTGGTTTGGTGTTGAACAAAGAGCACATCCGCAAGTTGGAACAAGACAAAGAATTACCACCCGACCTCGACCCTGAGGATCGTAGAGAACGTTACTTTGGATGGGATGGGTTAGTGAGATCGGGTGTGGTAGAGTATGTTGACGCCGAAGAAGAGGAGACGATCATGATTTCCATGACACCGGAAGACCTGGAAATTTCCAAACAGCTTCAGGCTGGCTACGCCCTTCCTGAAGAAGAGCTCGATCCCAACAAGCGCGTCCGCTCTATACTGAGCCAGAAGGCACATACTTGGACACACTGTGAGATTCATCCTAGTATGATTCTCGGAGTTTGTGCTAGTATCATTCCATTCCCTGATCACAATCAATCGCCCGTACCCAGGCCT
>11504cal
CCTCGAGGGAGCATCGTCGTTCGTGAAATTGGTTTTGTTAGTCGTCATGATTTGAACACAAGCTGACTTGGCTTTCTTGGGTTTCCTATAGGACAAGGACGGTGATGGTTAGTACAGTTTATTTTATTCATTCTCCCTTCAAATGCGACCAATATGTTTTAGCCGCCATAATTTTATCCAGTTTCTGTTCGATCGGCTGAAGTCTTGGCATTGATGAATTGACTTGATATGCAGGCCAGATCACCACCAAGGAGTTGGGCACTGTCATGCGCTCTCTGGGCCAAAACCCCTCTGAGTCGGAACTCCAGGACATGATTAACGAGGTTGACGCCGACAACAATGGCACCATTGACTTCCCTGGTACGAGACGGCTTCCGTACGATTCATAAATGAAATAGCTGTTAATGTTCAAATAGAGTTCCTGACGATGATGGCGAGAAAGATGAAGGATACCGACTCTGAGGAGGAGATCCGGGAGGCTTTCAAGGTTTTCGACCGCGATAACAACGGCTTCATCTCCGCTGCCGAATTGCGCCACGTTTGCCCCCCCTTTATCGGGATTT
>11504tub
GGAGGTCTCTGCCTTCGAGTTAGTATGCTTTGGACCAAGGAACTCCTCAAAAGCATGATCTCGGATGTGTCCTGTTATATCTGCCACATGTTTGCTAACAACTTTGCAGGCAAACCATCTCTGGCGAGCACGGCCTTGACGGCTCCGGTGTGTAAGTACAGCCTGTATACACCTCGAACGAACGACGACCATATGGCATTAGAAGTTGGAATGGATCTGACGGCAAGGATAGTTACAATGGCTCCTCCGATCTCCAGCTGGAGCGTATGAACGTCTACTTCAACGAGGTGCGTACCTCAAAATTTCAGCATCTATGAAAACGCTTTGCAACTCCTGACCGCTTCTCCAGGCCAGCGGAAACAAGTATGTCCCTCGTGCCGTCCTCGTTGATCTTGAGCCTGGTACCATGGACGCCGTCCGTGCCGGTCCCTTCGGTCAGCTCTTCCGTCCCGACAACTTCGTTTTCGGCCAGTCCGGTGCTGGTAACAACTGGGCCAAGGGTCATCACC
cf. Aspergillus TUCIM11505
>11505ITS
GCGTAGTACTGCGGAGGACATTACCGAGTGAGGGCCCTCTGGGTCCAACCTCCCACCCGTGTCTATCGTACCTTGTTGCTTCGGCGGGCCCGCCGTTTCGACGGCCGCCGGGGAGGCCTTGCGCCCCCGGGCCCGCGCCCGCCGAAGACCCCAACATGAACGCTGTTCTGAAAGTATGCAGTCTGAGTTGATTATCGTAATCAGTTAAAACTTTCAACAACGGATCTCTTGGTTCCGGCATCGATGAAGAACGCAGCGAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAGTCTTTGAACGCACATTGCGCCCCCTGGTATTCCGGGGGGCATGCCTGTCCGAGCGTCATTGCTGCCCTCAAGCACGGCTTGTGTGTTGGGCCCCCGTCCCCCTCTCCCGGGGGACGGGCCCGAAAGGCAGCGGCGGCACCGCGTCCGGTCCTCGAGCGTATGGGGCTTTGTCACCTGCTCTGTAGGCCCGGCCGGCGCCAGCCGACACCCAACTTTATTTTTCTAAGGTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAAGCATATCAATAAGCGGAGGAAAAAAAAACCCCACCCAATTTAA
>11505rpb2
AGAATCAGCCGTGTCTCGCAGGTACTGTCTCGTTACACCTATGCCTCCACCTTGTCACATCTTCGGCGTACCAACACGCCAATTGGTCGTGACGGTAAGATCGCAAAGCCTCGTCAGCTTCACAACACCCATTGGGGCTTGGTCTGTCCTGCAGAAACTCCTGAAGGTCAGGCTTGTGGTCTGGTCAAGAACCTGGCTCTCATGTGTTATATCACTGTCGGCACACCCAGCGAGCCCATCATCGATTTCATGATTCAGCGCAACATGGAAGTGCTTGAGGAGTTTGAGCCTCAGGTGACACCCAATGCAACGAAGGTGTTTGTGAACGGTGTTTGGGTCGGTGTCCACCGAGATCCCGCGCACTTGGTCAACACCATGCTCTCTCTGCGTCGGCGCAACATGATCTCTCACGAGGTCAGCTTGATTCGGGACATTCGTGAGCGTGAATTCAAGATCTTCACAGACGCTGGGCGTGTGTGTCGACCACTTTTCGTCGTTGACAATGACCCTAAGAGCGAGAACTGCGGCTCGCTGGTGCTCAACAAGGATCATATCCGCAAGCTAGAGCAAGATAAAGAACTACCCGCGGACCTTGATCCCGAAGATCGCAGGGAACGTTATTTCGGTTGGGATGGTTTGGTCAAGTCAGGAGTTGTCGAGTATGTCGATGCCGAGGAAGAAGAGACGATTATGATCGTTATGACCCCCGAAGACCTCGAAATCTCCAAACAGCTTCAGGCGGGTTACGCTCTTCCGGAAGAAGACGATCCCAACAAGCGTGTTCGTTCAGTCCTCAGTCAAAAGGCACACACTTGGACGCATTGCGAGATTCACCCCAGTATGATTCTTGGTGTTTGTGCCAGTATCATTCCCTTCCCGGACCACAACCAGTCGCCTCGTATCCTGCCCCC
>11505cal
GAGGGGACTGTCCAGTCCCTGGTCGTTGTATAGGAGGGATCTCCAGAATATTGAGGGTGTGCGCTGACACGAGATTTGACGTATAGGACAAGGATGGTGATGGTTAGTGACCCTTTTTCCACTCCTCGAACTTCGGCTTCCATGCGATCATGTTCAAACGCCGACTCACAATATCCGGAAATGACCCGTCAGTACTGATAATATCTATGTTTGACTATCAGGCCAGATCACCACCAAGGAATTGGGCACTGTAATGCGCTCTCTGGGCCAGAACCCTTCCGAGTCAGAGCTGCAAGATATGATCAACGAGGTGGATGCTGACAACAACGGCACCATCGATTTCCCCGGTATGTGATACTTTCGGTATGAACTCGGGAGGGGAGAGAACAATCATTAACTTGTAATCAGAATTCCTTACCATGATGGCTCGGAAGATGAAGGACACCGACTCCGAAGAGGAAATTCGGGAAGCTTTCAAGGTCTTCGACCGCGACAACAACGGTTTCATCCTCCGCTGCGGAGCTGCGCCACGTTGCCCCCCCTTTTACGGGGAACGC
>11505tub
GGGCGTCTTGACTCAGCTTGGATGACGGGTGATTGGGATCTCTCATCTTAGCAGGCTACCTCCATGGGTTCAGCCTCACTGTCATGGGTATCAGCTAACAAATCTACAGGCAGACCATCTCTGGTGAGCATGGCCTTGACGGCTCTGGCCAGTAAGTTCGACCTATATCCTCCCAATTGAGAAAGCGGCGGAAACACGGAAAACAAGGAAGAAGCGGACGCGTGTCTGATGGGAAATAATAGCTACAATGGCTCCTCCGATCTCCAGCTGGAGCGTATGAACGTCTATTTCAACGAGGTGTGTGGATGAAACTCTTGATTTATACTATTTCGGCAACATCTCACGATCTGACTCGCTACTAGGCCAACGGTGACAAATATGTTCCTCGTGCCGTTCTGGTCGATCTCGAGCCTGGTACCATGGACGCTGTCCGTGCCGGTCCCTTCGGCGAGCTATTCCGTCCCGACAACTTCGTCTTCGGCCAGTCCGGTGCTGGTAACAACTGGGCCAAGGGTCAAA
cf. Aspergillus TUCIM11502
>11502ITS
AGGGTGACTGCGGAGGATCATTACCGAGTGCGGGCTGCCTCCGGGCGCCCAACCTCCCACCCGTGACTACCTAACACTGTTGCTTCGGCGGGGAGCCCCCCAGGGGCGAGCCGCCGGGGACCACTGAACTTCATGCCTGAGAGTGATGCAGTCTGAGCCTGAATACAAATCAGTCAAAACTTTCAACAATGGATCTCTTGGTTCCGGCATCGATGAAGAACGCAGCGAACTGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAGTCTTTGAACGCACATTGCGCCCCCTGGCATTCCGGGGGGCATGCCTGTCCGAGCGTCATTGCTGCCCTCAAGCCCGGCTTGTGTGTTGGGTCGTCGTCCCCCCCGGGGGACGGGCCCGAAAGGCAGCGGCGGCACCGTGTCCGGTCCTCGAGCGTATGGGGCTTTGTCACCCGCTCGATTAGGGCCGGCCGGGCGCCAGCCGGCGTCTCCAACCTTATTTTTCTCAGGTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAAGCATATCAATAAGCGAGAAAAAAAAACCCCCCCCCCAAAACCCCCC
>11502rpb2
GAGCAGCGGCGGATGTGTCGCAGTGCTCAGTCGCTACACCTACGCTTCAACATTATCTCATCTTCGGCGAACAAACACGCCTATTGGTCGTGACGGCAAGATTGCTAAACCTCGTCAGCTACACAACACGCATTGGGGTCTGGTATGTCCTGCGGAAACCCCTGAAGGCCAAGCTTGCGGTCTGGTCAAGAACTTGGCTCTTATGTGCTACATTACTGTTGGTACTCCTAGCGAACCCATCATCGATTTCATGATTCAGCGTAACATGGAAGTGCTTGAGGAGTTTGAGCCTCAGGTAACACCGAACGCCACTAAGGTCTTTGTCAATGGCGTCTGGGTTGGTGTCCACCGTCAGCCTTCTCACCTTGTTGAGACCATGCAAGCCCTTCGTCGGCGAAACATGATTTCTCACGAAGTCAGTCTTATTCGAGACATCCGTGAACGAGAATTCAAGATCTTCACCGACGCCGGTCGTGTATGCCGGCCACTCTTCGTCGTTGACAACGATCCCAAGAGTGAGAACTGCGGCAACCTAGTCCTCAACAAGGAGCACATTCGCAAGTTGGAAGAAGACAAGGAACTTCCACCGGATATGGACCCAGAAGACCGACGGGAGCGCTACTTTGGATGGGACGGTCTCGTCAAATCCGGTGTCGTTGAGTACGTAGATGCCGAAGAGGAGGAGACGATCATGATCGTTATGACGCCCGAGGACTTGGAGATTTCAAAGCAGCTGCAGGCTGGTTACGCACTTCCGGAAGAGCAAGATCCAAACAAGCGTGTGCGGTCGATTCTCAGCCAGAGGGCGCACACCTGGACACACTGCGAGATTCACCCCAGTATGATTCTTGGTGTCTGCGCCAGTATTATCCCCTCCCAGATCACAACCAGTCTCCTCGTACACGTCCCC
>11502cal
CTTGGAAGTGCATTGGTTACTGTTATATCAAAATCGAATTTGTATTGAGAGTATACTAATACATTCCGCACTAAACAGGACAAGGATGGCGATGGTTAGTGCATCTGTCCCCCCAGGCTTGATCGCATTCGCCCAGCATGTCTGCTGTAGCTCTATATAACCGTTTCTGACAAACGGCGACAGGCCAGATTACCACTAAGGAGCTTGGCACTGTCATGCGCTCGCTCGGTCAGAATCCTTCAGAGTCTGAGCTTCAGGACATGATCAACGAAGTTGACGCCGACAACAATGGCACCATTGACTTTCCAGGTACGCGAACTCCCCAATCTACTTCGCACCAGCCTAGAAATGTACTAATGCTAAACAGAGTTCCTTACCATGATGGCCAGAAAGATGAAGGACACCGATTCCGAGGAGGAAATTCGGGAGGCGTTCAAGGTCTTCGACCGTGACAACAATGGTTTCATCTCCGCTGCTGAGCTGCGTCACGTTATGCCCTCTATCGG
>11502tub
CTATGTCGCCCGGACTGGCCGAAACGAAGTTGTCGGGACGGAAGAGCTCGCCGAAGGGACCGGCGCGGACGGCATCCATAGTACCGGGCTCGAGATCGACGAGGACGGCACGGGGAACGTACTTGTTACCGCTGGCCTGGACGAAGTCAGACACACCAAACTGCTACCTGATATCTCATCCACTGACCTCATTGAAGTAGACGTTCATACGCTCGAGTTGAAGGTCGGAGGTACCATTGTAACTGGGTTGTGTCAGGTTCTGTTGAGGTGTCGTGTTTTCAGCGGATGCTGGACGGATTGTACTCACACACCGGAGCCATCGAGGCCGTGCTCACCGGAGATGGTCTGCCTGTTGGCTTGTTAGCAGGTGTACGAAGAAAGAAATCATTCCAAAAAGGTGAAATTTTCGCTCACCAGAAAGCAGCACTTTT
cf. Cladosporium TUCIM11509
>11509ITS
GCGAGTACTGCGGAGGACATTACAAGTGACCCCGGTCTAACCACCGGGATGTTCATAACCCTTTGTTGTCCGACTCTGTTGCCTCCGGGGCGACCCTGCCTTCGGGCGGGGGCTCCGGGTGGACACTTCAAACTCTTGCGTAACTTTGCAGTCTGAGTAAACTTAATTAATAAATTAAAACTTTTAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCGAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCCCTGGTATTCCGGGGGGCATGCCTGTTCGAGCGTCATTTCACCACTCAAGCCTCGCTTGGTATTGGGCATCGCGGTCCGCCGCGTGCCTCAAATCGACCGGCTGGGTCTTCTGTCCCCTAAGCGTTGTGGAAACTATTCGCTAAAGGGTGTTCGGGAGGCTACGCCGTAAAACAACCCCATTTCTAAGGTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAAGCATATCAATAAGCGGAGGAAAGAAAACCCAACCAAATAATA
>11509tef1
ACTCGGGAATGCACCTTGTCGCGATGGCATATCCCTCTGCCCCGCCACAACACCCCGCCTCGTCGCAAATTTGCGATAAGGATGTGGGCCGGCCTGGCTTGGCATGGAGATGCATCGCGACAAGCCAGCATCGCCACTCAATACGCCTCGTTGAGCACACCACTGACAATCGCAATAGGAAGCCGCCGAACTCGGTAAGGTTCCCTTCAAGTAA
>11509act
GGGGTATTCGAAGAGCCGTTTTCCGTAAGTCTGAAGACACCTGTTTCGCCCATCTCGCAATTCCGAGCTGACACCCCTCCCAGCTTCCATTGTCGGCAGACCCCGTCACCATGGGTATGCATTCTTCCCCGCGAGCCTCCCTGTCGCGCGCAGCCAATTCTAACCCCTCCGCAGTATCATGATCGGTATGGGCCAGAAAGACTCGTAAAATTTTTTATAATGT
cf. Cladosporium TUCIM11510
>11510ITS
GCGAGTACTGCGGAGGACATTACAGTGACCCCGGTTTACCACCGGGATGTTCATAACCCTTTGTTGTCCGACTCTGTTGCCTCCGGGGCGACCCTGCCTTCGGGCGGGGGCTCCGGGTGGACACTTCAAACTCTTGCGTAACTTTGCAGTCTGAGTAAACTTAATTAATAAATTAAAACTTTTAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCGAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCCCTGGTATTCCGGGGGGCATGCCTGTTCGAGCGTCATTTCACCACTCAAGCCTCGCTTGGTATTGGGCAACGCGGTCCGCCGCGTGCCTCAAATCGACCGGCTGGGTCTTCTGTCCCCTAAGCGTTGTGGAAACTATTCGCTAAAGGGTGCTCGGGAGGCTACGCCGTAAAACAACCCCATTTCTAAGGTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAAGCATATCAATAAGCGGAGGAAAGAAAAACCCAACCAAAAAAA
>11510tef1
CAGTCTTCGGCATGGCGTCCTCTTCGCAGTGTTGTACCCCTCCGCCCCACCGCGCGCCACCCCACCTCGTCGCAATCTGCGATAAGATGTGGGACACGACTTGGCCGGGCACGGACAACACTGCGATGAAGACTTCATCGCCACACGCCATCACCATCACAATACATTGCTAACAACCCTCCCAGGAAGCCGCTGAACTCGGTAAGGTTCCCTTCAAGT
>11510act
TATGTGCAAGGCCGGTTTCGCCGGTGACGATGCGCCCAGAGCCGTTTTCCGTAAGTTTGCAGACACCTGTTTCGCCGAGCTCAGAACATCCAGCTGACAGTCTCTTAGCTTCCATTGTCGGTCGTCCCCGTCACCATGGGTATGCACTCCTCCCGCAAGCTCCGCCCTCGAGCCCAGATGTCTAACAGCAGCGCAGTATCATGATCGGTATGGGCCAGAAGGACTCGTAA
cf. Colletotrichum TUCIM11523
>11523ITS
TTTGCCTAGCCTGATCCGAGGTCAACCTGTAAAGAATTTGGGGGTTTAACGGCAAGAGTCCCTCCGGATCCCAGTGCGAGACGTTAGTTACTACGCAAAGGAGGCTCCGGGAGGGTCCGCCACTACCTTTAAGGGCCCACGTCGGCCGTGGGGCCCCAAAACCAAGCGGTGCTTGAGGGTTGAAATGACGCTCGAACAGGCATGCTCGCCAGAATGCTGGCGAGCGCAATGTGCGTTCAAAGATTCGATGATTCACTGAATTCTGCAATTCACATTACTTATCGCATTTCGCTGCGTTCTTCATCGATGCCAGAACCAAGAGATCCGTTGTTAAAAGTTTTAATTATTTGCTTGTGCCACTCAGAAGAGACGTCGTGTAAATAGAGTTTGGTTTCCTCCGGCGGGCGCCCCGTCCCCGTGGTGGGGGCCGGCGCCGGGAGGGGAGGCCCGCGAGAGGCTTCCCCTGCCCGCCGAAGCAACGGTTAGGTACGTTCACAAAGGGTTATAGAGCGGTAACTCAGTAATGATCCCTCCGCTGGTTCACCAACGGAGACCTTGTCGAA
>11523tub
ACTCCCTCATTCTGCGACGCGTCGGAATGGCCACTATTTCGGGATAGCCCCTGAGCGTACCCCGCCGATATTCCCACCCAAAACGACATTTACCAAACATGGCCCGACTTGCATCTCCAACGCTTCTAGCATGGGAAACATTCCGCTGACCATTGATTGTTTTCTCGCAATAGGTTCACCTTCAGACCGGCCAGTGCGTAAGTATCTCCTGATCTCAACACAACGAGCCAGAGTGCGGGGCTAACTTCTTTGAACAGGGTAACCAGATTGGTGCCGCCTTCTGGTGCGTAGCCAACCGCTCACGACGCGGCGATTTCGACATTTGATACGATTTCGTACTGACCTTGGTACAGGCAAAACATCTCTGGCGAGCACGGTCTCGACAGCAATGGCGTGTATGTCACTTTGTCCTCCAGTGCGGCTGCCCCATGGACCTAGTAGCTAATTATACCACAGGTACAACGGCACTTCCGAGCTCCAGCTCGAGCGTATGAGCGTCTACTTCAACGAAGTTTGTTATCCTAGTCCCCCAGTGTGCAGGCAATCCTATTGACGAATGCTGACCTTCTCACCCAACCAGGCCTCCGGCAACAAGTACGTTCCCCGCGCCGTCCTCGTCGACTTGGAGCCCGGTACCATGGACGCCGTCCGTGCCGGTCCCTTTGGCCAGCTTTTCCGCCCCGACAACTTCGTCTTTGGCCAGTCCGGTGCCGGCAACAACTGGGCCAAGGGTCACTACACCGAGGGAGCTGAGCTTGTCGACCAGGTTCTCGATGTCGTCCGTCGCGAGGCCGAGGGCTGCGACTGCCTCCAGGGCTTCCAGATCACCCACTCTCTTGGTGGTGGTACCGGTGCCGGTATGGGTACCCTCCTGATTTCCAAGATCCGTGAGGAGTTCCCCGACCGCATGATGGCCACCTTCTCCGTCGTTCCCTCTCCCAAGGTTTCCGACACCGTCGTCGAGCCCTACAACGCCACTCTCTCCGTCCACCAGCTGGTCGAGAACTCCGACGAGACCTTCTGCATTGACAACGAGGCTCTCTACGACATCTGCATGCGTACCCTCAAGCTCTCCAACCCCTCTTACGGCGACCTGAACCACCTCGTCTCCGCCGTCATGTCCGGTGTCACCACCTGCCTCCGTTTCCCCGGTCAGCTGAACTCTGACCTGCGCAAGCTCGCCGTCAACATGGTTCCTTTCCCCCGTCTCCACTTCTTCATGGTCGGCTTCGCTCCCCTGACCAGCCGTGGCGCCCACTCCTTCCGCGCTGTCAGCGTTCCCGAGCTCACCCAGCAGATGTTCGACCCCAAGAACATGATGGCCGCCTCTGACTTCCGCAACGGTCGTTACCTGACCTGCTCTGCCATCTTGTAAGTTGACTACAATGCCCCTAGAGTGCGAATGAATGCTAACTTTGTTGTTCCCAGCCGTGGTAAGGTCGCCATGAAGGATGTCGAGGACCAGATGCGCAATTCCAGAACAAGAACTCGTCCA
>11523act
TCGGGAAGCGCGAGCTGTCTTCCGTAAGTTCCCCCTCATCCGCAGACCGCAATCTTCTCCATAAGGGGTATACGATTTCGGTCATCCATACTCGAGATTGTTGATTCTAACTGTCCCTTAGCCTCCATTGTCGGTCGCCCCCGTCACCATGGGTAGGTCTATCTCTTGCTACGGCAATGCCATCTCCCGCGCCGCGACCTAACACGTGCACAGTATCATGATTGGTATGGGCCAGGAGGACTCGTA
cf. Fusarium TUCIM11349
>11349ITS
GAGGACGCGGAGGGACATTACCGAGTTTACAACTCCCAAACCCCTGTGAACATACCTTATGTTGCCTCGGCGGATCAGCCCGCGCCCCGTAAAAAGGGACGGCCCGCCGCAGGAACCCTAAACTCTGTTTTTAGTGGAACTTCTGAGTATAAAAAACAAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCAAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCGCCAGTATTCTGGCGGGCATGCCTGTTCGAGCGTCATTTCAACCCTCAAGCCCAGCTTGGTGTTGGGAGCTGCAGTCCTGCTGCACTCCCCAAATACATTGGCGGTCACGTCGAGCTTCCATAGCGTAGTAATTTACACATCGTTACTGGTAATCGTCGCGGCCACGCCGTTAAACCCCAACTTCTGAATGTTGACCTCGGATCAGGTAGGAATACCCGCTGAACTTAAGCATATCAATAAGCGGAGGAAAAAAAAACCAACCAAAGGGGGGGGGGGG
>11349rpb2
GCGTGAGGATGGTGTCTCAGGTGCTGACAGATATACATTTGCTTCCACTCTCTCGCATTTGCGACGTACCAATACGCCCATTGGACGTGATGGTAAGCTTGCGAAACCTCGCCAGCTGCACAACACTCACTGGGGCTTGGTGTGCCCTGCTGAGACGCCTGAAGGTCAGGCTTGTGGTCTGGTCAAGAACTTGTCGCTTATGTGCTATGTAAGCGTGGGCTCACCAGGCGAGCCTTTGATCGATTTTATGGTCAGCCGAGGTATGGAAGTCGTTGAGGAGTACGAGCCGACAAGATACCCTCACGCAACCAAGGTTTTCGTTAACGGTAGCTGGGTCGGTGTTCACCCTGAGCCTAGGGCATTGGTGAACAGCGTTTTGGAAACACGACGAAAGTCTTATCTCCAGTTCGAAGTCTCTCTTGTTCGTGATATCAGAGACCGCGAATTCAAGATCTTCTCCGACGCAGGCCGCGTCATGAGACCAGTCTTTACAGTGCAACAAGAGGATGACTACGAGACTGGCCTTACTAAAGGACAGCTAGTATTGACAAAGGATCTCGTGAATAAGATTGCCCAAGAGCAAGCAGAGCCACCTTCCGATCCATCAGAGAGGATTGGTTGGCAGGGACTGATTCGTTCTGGAGCTGTCGAGTACATTGATGCCGAAGAGGAGGAGACTGCTATGATTTGCATGACGCCTGAAGATCTCGAAATCTATCGTGAACAGAAGCGTGATGAAGTCAACCTAACAGAAGAAGAGAAGCAGGCCAAGTTGGAGGCAGAGAAGAGGGAACAGGAGGAAGAGCGCAACAAGCGCTTAAAGACAAAGGTCAACCCGACAACTCACATGTACACGCATTGTGAGATTCATCCCAGTATGATTCTGGGCATTTGTGCCAGTATCATTCCTTTCCCCGATCACAACCAGGTATGTGTCATCCCCCTTGATTGAGTTGAGTGCGACTAACAACTGCAGTCTCCTCGTACACATCCTTC
>11349tef1
CCCCCCTCTTTTGGGGGGGGGGGGCCCCCCCCCGCAGTCTGATGAAATCACGGTGACCGGGAGCGTCTGATAGCCATGTTAGTATGAGAATGTGATGACAGCAGTGGTGACAACATACCAATGACGGTGACATAGTAGCGAGGAGTCTCGAACTTCCAGAGGGCGATATCAATGGTGATACCACGCTCACGCTCGGCTTTGAGCTTGTCAAGAACCCAGGCGTACTTGAAGGAACCCTTACCGAGCTCGGCGGCTTCCTATTGACAGGTGGTTAGTGACTGGTTGACACGTGATGATGAGCGCCCAGGGAATGGTTTGTGGGAAGAGGGCAAACGCCTGTCGCTCGAGTGGCGGGGTATGAGCCCCACCAGGGAAAAAAATTACGACAAAGCCGCAAAATTTTTGACCTCGAGCGGGGTAACAGGCGCGTATCGAGTCGTCGTGTGAGTGCGATTCGAATGATATTTCGAAAGGAAAAGGGCGCGCGATCGAGGAAAATGAGACCAACCTTCTCGAACTTCTCGATGGTTCGCTTGTCGATACCACCGCACTGGTAGATCAAGTGACCGGTCTATCAAAGTATGTCAGCACATTGGAAATTTGAAACTACCCCGCCAAGTGTCGGCGGGGTTGGGATGCGGTGGTACTCACAGTGGTCGACTTGCCAGAGTCGACGTGGCCGATGACGACGACGTTAAGGTAGCTCCCCCCTCCCCCCAAAAAAAAAAAGAAACGGGCTGA
cf. Fusarium TUCIM11437
>11437ITS
TTGGTACGGCGGAGGGTCATTACCGAGTTTACAACTCCCAAACCCCTGTGAACATACCAATTGTTGCCTCGGCGGATCAGCCCGCTCCCGGTAAAACGGGACGGCCCGCCAGAGGACCCCTAAACTCTGTTTCTATATGTAACTTCTGAGTAAAACCATAAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCAAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCGCCAGTATTCTGGCGGGCATGCCTGTTCGAGCGTCATTTCAACCCTCAAGCCCTCGGGTTTGGTGTTGGGGATCGGCGAGCCCTTGCGGCAAGCCGGCCCCGAAATCTAGTGGCGGTCTCGCTGCAGCTTCCATTGCGTAGTAGTAAAACCCTCGCAACTGGTACGCGGCGCGGCCAAGCCGTTAAACCCCCAACTTCTGAATGTTGACCTCGGATCAGGTAGGAATACCCGCTGAACTTAAGCATATCAATAAGCGAGAAAAAAACCCACCCCCCCAAAAA
>11437rpb2
TTGACAGATATACCTTCGCCTCTACTCTTTCACATTTGCGCCGAACCAATACTCCCATTGGACGAGATGGTAAATTGGCCAAGCCTCGACAGCTTCACAATACTCACTGGGGTTTGGTGTGTCCCGCCGAAACGCCTGAGGGTCAAGCTTGTGGTCTGGTCAAAAACTTGTCTCTGATGTGTTATGTCAGTGTCGGCTCTCCAGCCGATCCTCTCATTGAATTCATGATCAACAGAGGTATGGAAGTCGTTGAGGAGTACGAGCCCACAAGATATCCCCACGCTACAAAGATTTTCGTCAACGGTAGCTGGGTTGGTGTTCACGCCGACCCCAAGCATCTCGTGAATCAGGTTTTGGACACAAGACGAAAGTCGTACGTCCAGTTCGAAGTATCACTTGTTCGTGATATTCGAGACCGTGAATTCAAGATCTTTTCAGATGCTGGTCGTGTCATGAGACCTGTCTTCACAGTCCATCAGGAGGATGACTACGAGAACAACATCACCAAGGGACAACTAGTGTTGACAAAGGAACATGTCAACAGGCTAGCGCAAGAGCAGGCAGAGCCACCTGCCAACCCCGCTGACAAGTTTGGATGGGATGGCTTGATTCGCGAAGGAGCTGTCGAGTATCTCGACGCCGAGGAAGAAGAGACAGCCATGATTTGCATGACGCCAGAGGATCTCGAACTTTACCGTGAGCAAAAGAATGATGAAGCTACACTCACGGAAGAGGAGAAACGGGCCAAGGCTGAGGCAGAGAAGAGGGAACAGGAGGAGGACCGCAACAAGCGATTGAAGACAAAGGTCAACCCCACAACTCACATGTACACACATTGCGAGATTCATCCCAGTATGATTCTCGGTATCTGTGCCAGTATCATTCCTTTCCCCGATCACA
>11437tef1
AATTTGGAGGGTTCCCCCCGGTCATGTTCTTGATGAAATCACGGTGACCGGGAGCGTCTGAATGATGTGTTAGTATGAGGAAGTAGGATGAGGTATGAGCGACAACATACCAATGACGGTGACATAGTAGCGAGGAGTCTCGAACTTCCAGAGAGCAATATCGATGGTGATACCACGCTCACGCTCGGCCTTGAGCTTGTCAAGAACCCAGGCGTACTTGAAGGAACCCTTACCGAGCTCAGCGGCTTCCTATTGTCGAATGGTTAGTCGCTGCTTGACACGTGACAATGCGCTCATTGAGGTTGTGGACAGGAAAGGGTAAAAACGCGCCCATCGCTCGAGTGGCGGGGTAAATGCCCCACCAAAAAAATTACGGTCATATCGCAAAATTTTTGGTCTCGAGCGGGGTAGCAGGCACGTTTCGAATCGCAAGTGAAATCGGTGGGCAGAGGACGCGCGATCGAAGGGAAAGTGACTAACCTTCTCGAACTTCTCGATGGTTCGCTTGTCGATACCACCGCACTGGTAGATCAAGTGACCGGTCTGTGAAGCGATGTCAGCATGTTGTCTTCCAAGATGTACCCCGCCAGATCTTGGTCAGGATCACGATGGCAGATAAGCTCAACGTCCAGGGTAGTACTCACAGTGGTCGACTTGCCAGAGTCGACGTGGCCGATGACGACGACGTTAAGGTGAGTCTGCCCCCCTTTTTCCCCAAAAAAGTAAATTT
cf. Fusarium TUCIM11398
>11398ITS
CGTGGACGCGGAGGGACATTTCCGAGTTTACAACTCCCAAACCCCTGTGAACATACCTATACGTTGCCTCGGCGGATCAGCCCGCGCCCCGTAAAACGGGACGGCCCGCCCGAGGACCCTAAACTCTGTTTTTAGTGGAACTTCTGAGTAAAACAAACAAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCAAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCGCCAGTATTCTGGCGGGCATGCCTGTTCGAGCGTCATTTCAACCCTCAAGCTCAGCTTGGTGTTGGGACTCGCGGTAACCCGCGTTCCCCAAATCGATTGGCGGTCACGTCGAGCTTCCATAGCGTAGTAATCATACACCTCGTTACTGGTAATCGTCGCGGCCACGCCGTAAAACCCCAACTTCTGAATGTTGACCTCGGATCAGGTAGGAATACCCGCTGAACTTAAGCATATCAATAAGCGGAGGAAAAAAAACCCCCACCAAACC
>11398rpb2
GGTGGGCTGGTGTCTCTAGGTGTTGACAGATATACATTTGCTTCCACTCTTTCACATTTGCGACGTACCAACACGCCCATTGGTCGTGATGGTAAGCTTGCAAAGCCTCGACAGCTGCACAACACTCACTGGGGCTTGGTGTGCCCTGCCGAAACACCAGAAGGTCAGGCTTGTGGTCTGGTCAAGAACTTGTCGCTGATGTGCTACGTGAGTGTGGGTTCACCAGCCGAGCCTTTGATCGAGTTCATGATCAACAGAGGTATGGAAGTTGTTGAAGAATACGAACCAACAAGATATCCTCACGCAACCAAGGTTTTCGTCAACGGAAGTTGGGTCGGCGTCCACCCTGATCCTAGACACCTGGTGAACTCCGTCCTGGACACACGACGAAAATCTTACGTCCAGTTCGAAGTTTCCCTTGTTCGTGACATCCGTGACCGTGAGTTCAAGATTTTCTCTGATGCAGGCCGAGTCATGAGACCAGTCTTCACAGTGCAACAAGAGGATGATTACGAAACCGGCATCAACAAGGGACAACTAGTATTGACAAAGGAGCTCGTGAACAAGATTGCACAGGAGCAAGCAGAGCCACCTGCTGATCCATCGGAGAAGATTGGATGGGAGGGTCTGATCCGCTCTGGAGCTGTGGAGTATCTCGATGCCGAGGAAGAAGAAACCGCAATGATTTGTATGACGCCTGAGGATCTCGAAATCTATCGTGAACAGAAGCAGGATGAGGTTAATCTTACAGAGGAAGAGAAGCGGGCCAAGCAAGAAGCAGAGAAGAGGGAGCAAGAGGAAGAGCGCAACAAGCGTCTGAAGACGAAGGTCAATCCCACAACTCACATGTACACCCATTGTGAGATTCATCCCAGTATGATTCTGGGTATTTGTGCCAGTATCATTCCCTTCCCCGATCACAACCAGGTATGTATCCGCACCCCGATCAAGTTGAGCCTATCTAACGATTACAGTCTCCTCGAGCGGGA
>11398tef1
TCGGCTGACGGGAGCGTCTGGACATATGTTAGTGAAAGAAGGTAATGAATATGGGTGACGACAACATACCAATGACGGTGACATAGTAGCGAGGAGTCTCGAACTTCCAGAGGGCGATATCAATGGTGATACCACGCTCACGCTCGGCCTTGAGCTTGTCAAGAACCCAGGCGTACTTGAAGGAACCCTTACCGAGCTCGGCGGCTTCCTATTGTCGAATGGTTAGCTACTGATTGACACGTGATGCGCAAGATGATTTGTGGGAAGAGGGCAAACGCCTGTAACTCGAGTAAGCCCCACCAAAAATTTACGGTTCAACCGCAAAATTTGGTACTCGAGCGGGGTAACAGGCGAGTATTTAGTCGTCGTGAGACTAAGTCGAGTGATGGATCGGTGGGCAGAAGGCATGCGATCGAGGAAAATGGAAACCAACCTTCTCGAACTTCTCGATGGTTCGCTTGTCGATACCACCGCACTGGTAGATCAAGTGACCGGTCTATACAATCTTGTCAGCAAACATTCAGCTTGAAACTACCCCGCCAGGTATAGCGGGGTTGATGACTGCTGATAAGCAGATCATCGAGGGTAGTACTCACAGTGGTCGACTTGCCAGAGTCGACGTGGCCGATGACGACGACGTTAAGGTGAGTCTTTT
cf. Fusarium TUCIM11404
>11404ITS
GGTAACGCGTAGGGACATTACCGAGTTTACAACTCCCAAACCCCTGTGAACATACCTATACGTTGCCTCGGCGGATCAGCCCGCGCCCCGTAAAACGGGACGGCCCGCCCGAGGACCCCTAAACTCTGTTTTTAGTGGAACTTCTGAGTAAAACAAACAAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCAAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCGCCAGTATTCTGGCGGGCATGCCTGTTCGAGCGTCATTTCAACCCTCAAGCTCAGCTTGGTGTTGGGACTCGCGGTAACCCGCGTTCCCCAAATCGATTGGCGGTCACGTCGAGCTTCCATAGCGTAGTAATCATACACCTCGTTACTGGTAATCGTCGCGGCCACGCCGTTAAACCCCAACTTCTGAATGTTGACCTCGGATCAGGTAGGAATACCCGCTGAACTTAAGCATATCAATAAGCGAGAAAAAAAAAACCCCCCCAACAAAATGAGA
>11404rpb2
GGAAAAGGGCTGATGTCTCTAGTGTTGACAGATATACATTTGCTTCCACTCTTTCACATTTGCGACGTACCAACACACCTATTGGTCGTGATGGTAAGCTTGCGAAGCCTCGGCAGCTGCACAACACTCACTGGGGCTTGGTGTGCCCTGCCGAGACACCAGAAGGTCAGGCTTGTGGTCTGGTCAAGAACTTGTCGCTGATGTGCTACGTGAGTGTGGGTTCGCCAGCTGAGCCTTTGATCGAGTTCATGATCAACAGAGGTATGGAAGTTGTTGAAGAATACGAACCAACAAGATATCCTCACGCAACCAAGGTCTTCGTCAACGGAAGTTGGGTCGGCGTCCACCCTGATCCCAGGCACCTGGTGAACTCCGTCCTGGACACACGACGAAAGTCTTACGTCCAGTTCGAGGTTTCCCTTGTTCGTGACATCCGTGACCGTGAATTCAAAATTTTCTCTGATGCAGGCCGAGTCATGAGACCAGTTTTCACAGTGCAGCAAGAGGATGATTACGAAACCGGCATCAATAAGGGACAGCTAGTATTGACAAAGGAGCTCGTGAACAAGATTGCCCAGGAGCAGGCAGAGCCACCTGCTGATCCATCGGAGAAGATTGGATGGGAGGGCCTCATCCGCTCTGGAGCTGTGGAGTATCTCGACGCCGAGGAAGAAGAAACCGCAATGATTTGCATGACGCCTGAAGATCTCGAAATCTACCGTGAACAGAAGCAGGACGAGGTTAATCTTACAGAGGAAGAGAAGCGGGCCAAGCAAGAAGCAGAGAAGAGGGAGCAAGAGGAGGAACGCAACAAGCGTCTGAAGACGAAGGTCAACCCCACAACTCACATGTACACACACTGTGAGATTCATCCCAGTATGATTCTGGGTATTTGTGCCAGTATCATTCCCTTCCCCGATCACAACCAGGTATGTATCCCCACGCCCGACCAAGTTGAGCCTGTCTAACGATTGCAGTCTCCTCGACACAGACTCC
>11404tef1
GTGCCCCCCTCACATTTTGAGAAATCACGGTGACCGGGAGCGTCTGAAGCACATGTTAGCATGAGGGAAATGATGGGTGTAAGTGATGACAACATACCAATGACGGTGACATAGTAGCGAGGAGTCTCGAACTTCCAGAGGGCGATATCGATGGTGATACCACGCTCACGCTCGGCCTTGAGCTTGTCAAGAACCCAGGCGTACTTGAAGGAACCCTTACCGAGCTCGGCGGCTTCCTATTGTCGGGTGGTTAGTGACTGGTTGACACATGATGCGCGAGACATGGATTTGTGGGAAGGGCAAACGCCTGTCACTCGAGTAGCGGGGTTGAAACCCCACCAAAAGAATTACGGTTGAGCCGCAAAATTTTGTACTCGAGCGGGGTAACAGGCACATATTCAATCGTCGTAAAACTGATTCGACTGATAGATCGGAGGGTAGAGGGCGTGCGATCGAGGGAAATGGGAACCAACCTTCTCGAACTTCTCGATGGTTCGCTTGTCGATACCACCACACTGGTAGATCAAGTGACCGGTCTATGCAATCTTGTCAGCAGATATTCAAGTTAAAATTACCCCGCCACGTATGGCGGGGTTGATGACTGCTGATAAGCCGGTCATCGTGGGTAGTACTCACAGTGGTCGACTTGCCAGAGTCGACGTGGCCGATGACGACGACGTTAAGGTGATTGCCTCCCCCCCACAAAAAAAAAAAAAAGGAGGAGGCGGG
cf. Fusarium TUCIM11369
>11369ITS
TGGGGACGCGGAGGGTCATTACCGAGTTTACAACTCCCAAACCCCTGTGAACATACCTTATGTTGCCTCGGCGGATCAGCCCGCGCCCCGTAAAAAGGGACGGCCCGCCGCAGGAACCCTAAACTCTGTTTTTAGTGGAACTTCTGAGTATAAAAAACAAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCAAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCGCCAGTATTCTGGCGGGCATGCCTGTTCGAGCGTCATTTCAACCCTCAAGCCCAGCTTGGTGTTGGGAGCTGCAGTCCTGCTGCACTCCCCGAATACATTGGCGGTCACGTCGAGCTTCCATAGCGTAGTAATTTACACATCGTTACTGGTAATCGTCGCGGCCACGCCGTTAAACCCCAACTTCTGAATGTTGACCTCGGATCAGGTAGGAATACCCGCTGAACTTAAGCATATCAATAAGCGAGAAAAAAAACCCCCCCCCAAAAATAACCC
>11369rpb2
AGGGACGGCCTGTGTCTCAGGTGCTGACAGATATACATTTGCTTCCACTCTCTCGCATTTGCGACGTACCAATACGCCCATTGGACGTGATGGTAAGCTTGCGAAACCTCGCCAGCTGCACAACACTCACTGGGGCTTGGTGTGCCCTGCTGAGACGCCTGAAGGTCAGGCTTGTGGTCTGGTCAAGAACTTGTCGCTTATGTGCTATGTAAGCGTGGGCTCACCAGGCGAGCCTTTGATCGATTTTATGGTCAGCCGAGGTATGGAAGTCGTTGAGGAGTACGAGCCGACAAGATACCCTCACGCAACCAAGGTTTTCGTTAACGGTAGCTGGGTCGGTGTTCACCCTGAGCCTAGGGCATTGGTGAACAGCGTTTTGGAAACACGACGAAAGTCTTATCTCCAGTTCGAAGTCTCTCTTGTTCGTGATATCAGAGACCGCGAATTCAAGATCTTCTCCGACGCAGGCCGTGTCATGAGACCAGTCTTTACAGTGCAACAAGAGGATGACTACGAGACTGGCCTTACTAAAGGACAGCTAGTATTGACAAAGGATCTCGTGAATAAGATTGCCCAAGAGCAGGCAGAGCCACCTTCCGATCCATCAGAGAGGATTGGTTGGCAGGGACTGATTCGTTCTGGAGCTGTCGAGTACATTGATGCCGAAGAGGAGGAGACTGCTATGATTTGCATGACGCCTGAAGATCTCGAAATCTATCGTGAACAGAAGCGTGATGAAGTCAACCTAACAGAAGAAGAGAAGCAGGCCAAGTTGGAGGCAGAGAAGAGGGAACAGGAGGAAGAGCGCAACAAGCGCTTAAAGACAAAGGTCAATCCGACAACTCACATGTACACGCATTGTGAGATTCATCCCAGTATGATTCTGGGCATTTGTGCCAGTATCATTCCTTTCCCCGATCACAACCAGGTATGTGTCATCCCCCTTGATTAAGTTGAGCGCGACTAACAACTGCAGTCTCCTCGTACACATCCTC
>11369tef1
GGGCGCTTTTTGGGGGGGGGCCCCCCCCCCCGTAGTTCTTGATGAAATCACGGTGACCGGGAGCGTCTGATAGCCATGTTAGTATGAGAATGTGATGACAGCAGTGGTGACAACATACCAATGACGGTGACATAGTAGCGAGGAGTCTCGAACTTCCAGAGGGCGATATCAATGGTGATACCACGCTCACGCTCGGCTTTGAGCTTGTCAAGAACCCAGGCGTACTTGAAGGAACCCTTACCGAGCTCGGCGGCTTCCTATTGACAGGTGGTTAGTGACTGGTTGACACGTGATGATGAGCGCCCAGGGAATGGTTTGTGGGAAGAGGGCAGACGCCTGTCGCTCGAGTGGCGGGGTATGAGCCCCACCGGGAAAAAAATTACGACAAAGCCGCAAAATTTTTGACCTCGAGCGGGGTAACAGGCGCGTATCGAGTCGTCGTGTGAGGGCGATTCGAATGATATTTCGAAAGGGAAAAGGGCGCGCGATCGAGGAAAATGAGACCAACCTTCTCGAACTTCTCGATGGTTCGCTTGTCGATACCACCGCACTGGTAGATCAAGTGACCGGTCTATCAAAGTATGTCAGCACATTGGAAATTTGAAACTACCCCGCCAAGTGTCGGCGGGGTTGGGATGCGGTGGTACTCACAGTGGTCGACTTGCCAGAGTCGACGTGGCCGATGACGACGACGTTAAGGTGAGCTGCCCTCCCCCCTAAAAAAAAAAAAAAAAAGGGGGGG
cf. Fusarium TUCIM11367
>11367ITS
CGCGTAGGGTCATTACCGAGTTTACAACTCCCAAACCCCTGTGAACATACCTATACGTTGCCTCGGCGGATCAGCCCGCGCCCCGTAAAAAGGGACGGCCCGCCCGAGGACCCCTAAACTCTGTTTTTAGTGGAACTTCTGAGTAAAACAAACAAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCAAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCGCCAGTATTCTGGCGGGCATGCCTGTTCGAGCGTCATTTCAACCCTCAAGCTCAGCTTGGTGTTGGGACTCGCGGTAACCCGCGTTCCCCAAATCGATTGGCGGTCACGTCGAGCTTCCATAGCGTAGTAATCATACACCTCGTTACTGGTAATCGTCGCGGCCACGCCGTAAAACCCCAACTTCTGAATGTTGACCTCGGATCAGGTAGGAATACCCGCTGAACTTAAGCATATCAATAAGCGGAGGAAAGAAAAACCCCCCCCCAAAACAAAAA
>11367rpb2
GCTTCCACTCTTTCACATTTACGACGTACTAACACGCCTATTGGTCGTGATGGTAAGCTTGCGAAGCCTCGGCAGCTGCACAACACTCACTGGGGCTTGGTTTGCCCTGCCGAGACACCTGAAGGTCAGGCTTGTGGTCTGGTCAAGAACTTATCGCTGATGTGCTACGTGAGTGTGGGTTCACCAGCCGAGCCTTTGATCGAGTTCATGATCAACAGAGGCATGGAAGTTGTTGAAGAATATGAACCAACAAGATACCCTCACGCAACCAAGGTTTTCGTCAACGGAAGTTGGGTCGGCGTCCACCCTGATCCCAGGCACCTGGTGAATTCCGTCCTGGACACACGACGAAAATCGTATGTCCAGTTTGAAGTTTCCCTTGTTCGTGACATCCGTGATCGTGAATTCAAGATCTTCTCTGATGCAGGCCGAGTCATGAGACCAGTCTTCACAGTGCAACAAGAGGATGATTACGAAACCGGCATCAATAAGGGACAGCTAGTATTAACAAAGGAGCTCGTGAACAAGATTGCCCAGGAGCAAGCAGAGCCACCTGCTGATCCATCGGAGAAGATTGGATGGGAGGGCCTGATCCGCTCTGGAGCTGTGGAGTATCTCGACGCCGAGGAAGAAGAAACCGCAATGATTTGCATGACGCCTGAAGATCTCGAAATCTATCGTGAACAGAAGCAGGACGAGGTTAATCTTACAGAGGAAGAGAAACGGGCCAAACAAGAAGCAGAGAAGAGAGAGCAAGAGGAAGAACGCAACAAGCGCCTGAAGACCAAGGTTAACCCCACAACTCACATGTACACACACTGTGAGATTCATCCCAGTATGATTCTGGGTATTTGTGCCAGTATTATTCCCTTCCCCGATCACA
>11367tef1
CCGGATCATGTTCTTGATGAAATCACGGTGACCGGGAGCGTCTGGAATACATGTTAGCCTGAGAAGGTAATGAGTGCAAGTGGTGACAACATACCAATGACGGTGACATAGTAGCGAGGAGTCTCGAACTTCCAGAGAGCGATATCGATGGTGATACCACGCTCACGCTCGGCCTTGAGCTTGTCAAGAACCCAGGCGTACTTGAAGGAACCCTTACCGAGCTCGGCGGCTTCCTATTGTCGAGTGGTTAGTGACTGATTGACACGTGATGCGCAAGTGATTTTGTGGGAAGAGGGCAAGCGCCTGTCACTCGAGTAGCGGGGTATAAGCCCCACCAAAAAATTACGGTTGAACCGCAAAATTTTGTACTCGAGCGGGGTAACAGGCGCATGTTCAGTCGTCGAGACTGATTCGGGTGCTGGATCGATGGGCAGAGGGCGTGCGATCGAGGAAAATGGAAACCAACCTTCTCGAACTTCTCGATGGTTCGCTTGTCGATACCACCGCACTGGTAGATCAAGTGACCGGTCTATGCAATTTTGTCAGCAAATGTGTAAGTTGAATTTACCCCACCACGTATGGCGGGGTTGATGACTGCTGATAAGCAGGTCATTGAGGGTAGTACTCACAGTGGTCGACTTGCCAGAGTCGACGTGGCCGATGACGACGACGTTAAGGTGAGTCTTGCTCCCTTTAACCCTTA
cf. Fusarium TUCIM11442
>11442ITS
TTGGATACGCGGAGGGTCATTACCGAGTTTACAACTCCCAAACCCCTGTGAACATACCTCTATGTTGCCTCGGCGGATCAGCCCGCGCCCCGTAAAACGGGACGGCCCGCCGCAGGAACCACAAACTCTGTTTTTAGTGGAACTTCTGAGTATAAAAAACAAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCAAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCGCCAGTATTCTGGCGGGCATGCCTGTTCGAGCGTCATTTCAACCCTCAAGCTCAGCTTGGTGTTGGGAGCTGCGGTCATCGCACTCCCCAAATTGATTGGCGGTCACGTCGAGCTTCCATAGCGTAGTAATTTACACATCGTTACTGGTAATCGTCGCGGCCACGCCGTTAAACCCCAACTTCTGAATGTTGACCTCGGATCAGGTAGGAATACCCGCTGAACTTAAGCATATCAATAAGCGGAGGAAGAAAAACCCCCACCAAAAAACTCC
>11442rpb2
GTTCATTTATTCCTGTTCTAGTGTGACAGATATACATTTGCCTCGACACTTTCGCATTTGCGACGTACCAACACGCCCATTGGACGTGACGGCAAGCTCGCGAAACCTCGCCAGCTGCACAACACTCACTGGGGCTTGGTGTGCCCTGCCGAGACGCCTGAAGGTCAGGCTTGTGGTCTGGTCAAGAATTTGTCGCTGATGTGCTATGTGAGTGTTGGTTCACCAGCTGAGCCTCTGATCGAATTCATGATCAACCGAGGCATGGAAGTCGTTGAGGAGTACGAACCAACAAGATACCCTCACGCAACCAAGGTTTTCGTTAACGGTAGCTGGGTCGGTGTCCACCCTGATCCTAAGGGACTGGTGAACAGCGTCTTGGATACGCGACGAAAGTCTTATGTCCAGTTTGAAGTCTCCCTTGTTCGTGATATTCGAGACCGTGAATTCAAAATCTTCTCTGACGCAGGCCGTGTCATGAGGCCAGTCTTCACCGTGCAACAAGAGGATGACTACGAAACTGGCATCAACAAAGGACAGCTAGTGTTGACAAAGGACCTTGTGAATAAGATTGCTCAGGAGCAAGCAGAGCCACCTTCTGATCCATCAGAGAAGATTGGTTGGGAGGGACTTATTCGCTCTGGAGCTGTCGAGTATCTCGATGCTGAAGAAGAGGAAACCGCTATGATTTGCATGACACCTGAAGATCTTGAAATCTATCGTGAACAGAAGCAGGACGAGATCAATCTTACAGAAGAAGAGAAGCAGGCCAAGCTGGAGGCAGAAAAGAGGGAACAAGAGGAAGAACGCAACAAGCGTTTGAAGACAAAGGTCAACCCCACGACTCACATGTACACACATTGTGAGATTCATCCCAGTATGATTCTGGGCATTTGTGCCAGTATCATTCCTTTCCCCGACCACAACCAGGTATGTGTTATCCCCCTTAACTAGGTCGAACGCGACTAACAATCGTAGTCTCCTCGTACACATCCTTTCC
>11442tef1
CCTTTTTGGGGGGGGCCCCCCCGGTCATGTTCTTGATGAAATCACGGTGACCAGGAGCGTCTTCAAGATGAGTTAGTATAGATATGTGATCAAGGTGATAGAGCCAACGTACCAATGACGGTGACATAGTAGCGAGGAGTCTCGAACTTCCAGAGAGCGATATCGATGGTGATACCACGCTCACGCTCGGCCTTGAGCTTGTCAAGAACCCAAGCGTACTTGAAAGAACCCTTACCGAGCTCGGCGGCTTCCTATTGACAGGTGGTTAGTGACTGATTGACACGTGATGATGCGCGCCCAGATGGATTGGGGAGGGCAAGAGCCTGTCGCTCGAGTGGTGGGGTATGAGCCCCACCAAAAAAATTACGACAAAATGGTAAAAATTTTGAACTCGAGCGGGGTAACAGGCGTGCGTCGGGTGGTCGTTTAAAAGTGATTCGGATGACGGATCGATATTTAGGGCGCGCGATCGAAGAAAATGAGACCAACCTTCTCGAACTTCTCGATGGTTCGCTTGTCGATACCACCGCACTGGTAGATCAAGTGACCGGTCTGTCCAGTATGTCAGCACAAGCTTATCTCGAGGCTATCCCGCCGAGTGTAGGCTGGGTTGAGGTTCAGTTGTACTTACAGTGGTCGACTTGCCAGAGTCGACGTGGCCGATGACGACGACGTTAAGGTGAGTCTGCCCCCCTTTTCCCCAAAAACCCA
cf. Fusarium TUCIM11370
>11370ITS
GGGAACGCGGAGGGACATTACCGAGTTTACAACTCCCAAACCCCTGTGAACATACCTATACGTTGCCTCGGCGGATCAGCCCGCGCCCCGTAAAACGGGACGGCCCGCCCGAGGACCCCTAAACTCTGTTTTTAGTGGAACTTCTGAGTAAAACAAACAAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCAAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCGCCAGTATTCTGGCGGGCATGCCTGTTCGAGCGTCATTTCAACCCTCAAGCTCAGCTTGGTGTTGGGACTCGCGGTAACCCGCGTTCCCCAAATCGATTGGCGGTCACGTCGAGCTTCCATAGCGTAGTAATCATACACCTCGTTACTGGTAATCGTCGCGGCCACGCCGTTAAACCCCAACTTCTGAATGTTGACCTCGGATCAGGTAGGAATACCCGCTGAACTTAAGCATATCAATAAGCGGAGAAGAAACAAAACCCCCAAAAAAAGTAA
>11370rpb2
ATGTCTCTAGTGTGACAGATATACATTTGCTTCCACTCTTTCACATTTGCGACGTACCAACACACCTATTGGTCGTGATGGTAAGCTTGCGAAGCCTCGGCAGCTGCACAACACTCACTGGGGCTTGGTGTGCCCTGCCGAGACACCAGAAGGTCAGGCTTGTGGTCTGGTCAAGAACTTATCGCTGATGTGCTATGTGAGTGTGGGTTCGCCAGCTGAGCCTTTGATCGAGTTCATGATCAACAGAGGTATGGAAGTTGTCGAAGAATACGAACCAACAAGATATCCTCACGCAACCAAGGTTTTCGTCAACGGAAGTTGGGTCGGCGTCCACCCTGATCCCAGGCACCTGGTGAACTCCGTCCTGGACACACGACGAAAGTCTTACGTCCAGTTCGAGGTTTCCCTTGTTCGTGACATCCGTGACCGTGAATTCAAGATTTTCTCTGATGCAGGCCGAGTCATGAGACCAGTCTTCACGGTGCAACAAGAGGATGATTACGAAACCGGCATCAATAAGGGACAGCTAGTATTGACAAAGGAGCTCGTGAACAAGATTGCCCAGGAGCAGGCAGAGCCACCTGCTGATCCATCGGAGAAGATTGGATGGGAGGGCCTCATCCGCTCTGGAGCTGTGGAGTATCTCGATGCCGAGGAAGAAGAAACCGCAATGATTTGCATGACGCCTGAAGATCTCGAAATCTATCGTGAACAGAAGCAGGACGAGGTTAATCTTACAGAGGAAGAGAAGCGGGCCAAGCAAGAAGCAGAGAAGAGGGAGCAAGAGGAAGAACGCAACAAGCGTCTGAAGACGAAGGTCAACCCCACAACTCACATGTACACACATTGTGAGATTCATCCCAGTATGATTCTGGGTATTTGTGCCAGTATCATTCCCTTCCCCGATCACAACCAGGTATGTATTCCCATCCCCGATCAAGTTGAGCCCATCTAACGACTACAGTCTCCTCGTACACTGAT
>11370tef1
CCCTTTATTTTTTGGGGGGGTTCCCCCCCCCCGTAGTCTGATGAAATCACGGTGACCGGGAGCGTCTGAAGTACATGTTAGCATGAGAAAGTATTGAGTGTAAGTGACGATAACGTACCAATGACGGTGACATAGTAGCGAGGAGTCTCGAACTTCCAGAGGGCGATATCGATGGTGATACCACGCTCACGCTCAGCCTTGAGCTTGTCAAGAACCCAGGCGTACTTGAAGGAACCCTTACCGAGCTCGGCGGCTTCCTATTGTCGGGTGGTTAGTGGCTGATGGACACGTGATGCACAAGACATGGGTTTCTGGGAAGAGGGCAAACGTCTGTCGCTCGAGTGGCGGGGTTGAAACCCCACCAAAAAAAATTACGGTTGAACCGCAAAATTTTGTACTCGAGCGGGGTAACAGGCGCATATTCAATCGTCGTAACTGATTCGACTGATGGATCGGTGGGTAGAGGGCGTGCGATCGGGGAAATGGAAACCAACCTTCTCGAACTTCTCGATGGTTCGCTTGTCGATACCACCGCACTGGTAGATCAAGTGACCGGTCTATGCAATCTTGTCAGCAAATATTCAAGTTGAAATTACCCCGCCACATCTGGCGGGGTTGATGACTGCTGATAAGCAAATCATCGTGGGTAGTACTCACAGTGGTCGACTTGCCAGAGTCGACGTGGCCGATGACGACGACGTTAAGGTGGCTGGCCTCTCCCCAAAAAAAAAAAAAAAGGGGCGTGGT
cf. Fusarium TUCIM11397
>11397ITS
CGTAATACGCGTGGGACATTACCGAGTTTACAACTCCCAAACCCCTGTGAACATACCTATACGTTGCCTCGGCGGATCAGCCCGCGCCCCGTAAAACGGGACGGCCCGCCCGAGGACCCCTAAACTCTGTTTTTAGTGGAACTTCTGAGTAAAACAAACAAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCAAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCGCCAGTATTCTGGCGGGCATGCCTGTTCGAGCGTCATTTCAACCCTCAAGCTCAGCTTGGTGTTGGGACTCGCGGTAACCCGCGTTCCCCAAATCGATTGGCGGTCACGTCGAGCTTCCATAGCGTAGTAATCATACACCTCGTTACTGGTAATCGTCGCGGCCACGCCGTAAAACCCCAACTTCTGAATGTTGACCTCGGATCAGGTAGGAATACCCGCTGAACTTAAGCATATCAATAAGCGAGGAAGAAAAACCCCCCCCAAACCT
>11397rpb2
GAGGAGGTCTGGGTGTCTCTAGTGTTGACAGATATACATTTGCTTCCACTCTTTCACATTTGCGACGTACCAACACACCTATTGGTCGTGATGGTAAGCTTGCGAAGCCTCGGCAGCTGCACAACACTCACTGGGGCTTGGTGTGCCCTGCCGAGACACCAGAAGGTCAGGCCTGTGGTCTGGTCAAGAACTTGTCGCTGATGTGCTACGTGAGTGTGGGTTCGCCAGCTGAGCCTTTGATCGAGTTCATGATCAACAGAGGTATGGAAGTTGTTGAAGAATACGAACCAACAAGATATCCTCACGCAACCAAGGTTTTCGTCAACGGAAGTTGGGTCGGCGTCCACCCTGATCCCAGGCACTTGGTGAACTCCGTCCTGGACACACGACGAAAGTCTTACGTCCAGTTCGAGGTTTCCCTTGTTCGTGACATCCGTGACCGTGAATTCAAGATTTTCTCTGATGCAGGCCGAGTCATGAGACCAGTCTTCACAGTGCAACAAGAGGATGATTACGAAACCGGCATCAACAAGGGACAGCTAGTATTGACAAAGGAGCTCGTGAACAAGATTGCCCAGGAGCAGGCAGAGCCACCTGCTGATCCATCGGAAAAGATTGGATGGGAGGGCCTCATCCGCTCTGGAGCTGTGGAGTATCTCGACGCCGAGGAAGAAGAAACCGCAATGATTTGCATGACGCCTGAAGATCTCGAAATCTATCGTGAACAGAAGCAGGATGAGGTTAATCTTACAGAGGAAGAGAAGCGGGCCAAGCAAGAAGCAGAGAAGAGGGAGCAAGAGGAAGAACGCAACAAGCGTCTGAAGACGAAGGTCAACCCCACAACTCACATGTACACACATTGTGAGATTCATCCCAGTATGATTCTGGGTATTTGTGCCAGTATCATTCCCTTCCCCGATCACAACCAGGTATGTATTCCCACCCCCAATCAAGTTGAGCCTATCTAACGACTACAGTCTCCTCGTACACTGATCCC
>11397tef1
GGAGCTCACCTTAACGTCGTCGTCATCGGCCACGTCGACTCTGGCAAGTCGACCACTGTGGAGTACCACCCTCGATAACCTGCTTATCAGCAGTCATTAACCCCGCCATATGTGGCGGGGTAATTTCAATAAACATCTGCTGACAAGATTGCATAGACCGGTCACTTGATCTACCAGTGCGGTGGTATCGACAAGCGAACCATCGAGAAGTTCGAGAAGGTTGGTTTCCATTTTCCTCGATCGCACGCCATCTACCCACCGATCCATCAGTCGAATCAGTCTTAGGACGATTGAATATGCGCCTGTTACCCCGCTCGAGTACAAAATTTTGCGGTACAACCGTATTTTTTTGGTGGGGTTTCAACCCCGCTACTCGAGCGACAGACGTTTGCCCTCTTCCCACAAACTCATGTCTTGCGCATCACGTGTCCATCAGTCACTAACCACCCGACAATAGGAAGCCGCCGAGCTCGGTAAGGGTTCCTTCAAGTACGCCTGGGTTCTTGACAAGCTCAAGGCTGAGCGTGAGCGTGGTATCACCATCGATATCGCCCTCTGGAAGTTCGAGACTCCTCGCTACTATGTCACCGTCATTGGTACGTTATCATCACTTACACTCAATACCTCCCTCATGCTAACATGTACTTCAGACGCTCCCGGTCACCGTGATTTCATCAAGACTGAGG
cf. Fusarium TUCIM11401
>11401ITS
GGGGGAGACGCGGAGGGACATTACCGAGTTTACAACTCCCAAACCCCTGTGAACATACCTTTATGTTGCCTCGGCGGATCAGCCCGCGCCCCGTAAAACGGGACGGCCCGCCGCAGGAAACCCTAAACTCTGTTTTTAGTGGAACTTCTGAGTATAAAAAACAAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCAAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCGCCAGTATTCTGGCGGGCATGCCTGTTCGAGCGTCATTTCAACCCTCAAGCCCAGCTTGGTGTTGGGATCTGTCTTCACTGACAGTCCTCAAATTGATTGGCGGTCACGTCGAGCTTCCATAGCGTAGTAATTTACACATCGTTACTGGTAATCGTCGCGGCCACGCCGTTAAACCCCAACTTCTGAATGTTGACCTCGGATCAGGTAGGAATACCCGCTGAACTTAAGCATATCAATAAGCGGAGGAAAAAAAACCCCAACCAAAAAA
>11401rpb2
GGAAGTATGTTTGGTGTCTCAGTGTGACAGATATACATTTGCCTCCACTCTTTCGCATTTGCGACGTACCAATACGCCCATTGGACGTGATGGTAAGCTTGCGAAGCCTCGCCAGCTGCACAACACTCACTGGGGCTTGGTCTGCCCTGCTGAGACGCCTGAGGGTCAGGCTTGTGGTCTGGTCAAGAACTTGTCGCTCATGTGTTATGTGAGTGTTGGTTCACCAGCCGAGCCTTTGATCGAGTTCATGATCAACAGAGGTATGGAAGTCGTTGAGGAATACGAGCCGACAAGATACCCTCACGCAACCAAGGTTTTCGTTAACGGTAGCTGGGTCGGTGTCCACCCTGATCCTAGGGGACTGGTGAACAGCGTCTTGGATACACGACGAAAGTCTTATGTCCAGTTTGAAGTCTCTCTTGTTCGTGATATTCGAGATCGTGAATTCAAGATCTTCTCTGACGCAGGCCGTGTCATGAGACCAGTTTTCACCGTGCAACAAGAGGACGACTACGAAACTGGCATCAATAAAGGACAGCTAGTACTGACAAAGGACCTCGTGAACAAGATTGCCCAGGAGCAAGCAGAACCACCTTCCGATCCATCAGAGAAGATTGGTTGGGAGGGACTGATTCGCTCTGGAGCTGTCGAGTACCTTGATGCTGAAGAAGAGGAAACCGCCATGATTTGCATGACGCCTGAAGATCTCGAAATTTACCGTGAACAGAAACAAGATGAAATCAACCTCACAGAAGAAGAGAAGCAGGCCAAGCTAGAGGCAGAGAAGAGGGAACAGGAAGAAGAACGCAACAAGCGTCTGAAGACAAAGGTTAACCCCACAACCCACATGTACACACATTGTGAGATTCATCCCAGTATGATTCTGGGTATTTGTGCCAGTATCATTCCCTTCCCTGATCACAACCAGGTATGTGCCATTTCCTCTGACTAAGTAAGTTGAATGCGCCTAACAATTTGCAGTCTCCTCGTACACCTACTTCC
>11401tef1
GCCTGGGTTTTTTTTTTTTTTTTTTTATGAAGAGGGGGAGCTCACCTTAACGTCGTCGTCATCGGCCACGTCGACTCTGGCAAGTCGACCACTGTGAGTAACTCTGCATCTTAACCCCGCCTAGATCTGGTGGGGTAGTTTCAATCATCGTTCTTACTGACATGCATCGATAGACCGGTCACTTGATCTACCAGTGCGGTGGTATCGACAAGCGAACCATCGAGAAGTTCGAGAAGGTTGGTCTCATTTTCCTCGATCGCGCGCCCTTCTTCCCATCGATCCGTCATTCGAATCGCTCTTCTACGACGACTCGACAAGCGCCTATCATTACCCCGCTCGAGTTCAAAGTTTTTTACGGTTTCGTCGTAATTCTTTTTGGTGGTGGGGCTTATACCCCGCCACTCGAATGACAGGCGCTTCCCCTTCCCACAGCCATTTTTATGGGCGCGCACCATCACGTGTCAATCAGTCACTAACCACTCGTCAATAGGAAGCCGCCGAGCTCGGTAAGGGTTCCTTCAAGTACGCCTGGGTTCTTGACAAGCTCAAAGCCGAGCGTGAGCGTGGTATCACCATTGATATCGCTCTCTGGAAGTTCGAGACTCCTCGCTACTATGTCACCGTCATTGGTATGTCGCCACCCTCGTCTTCATCACACTCTCATACTAACATGACTATTAGACGCTCCCGGTCACCGTGATTTCATCAAGACTGCGGGGGGGGGCCCCCCAAAAAAAAAAAAAACAACGCCCAATC
cf. Fusarium TUCIM11467
>11467ITS
GGGGGACGCGTAGGGACATTACCGAGTTTACAACTCCCAAACCCCTGTGAACATACCACTTGTTGCCTCGGCGGATCAGCCCGCTCCCGGTAAAACGGGACGGCCCGCCAGAGGACCCCTAAACTCTGTTTCTATATGTAACTTCTGAGTAAAACCATAAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCAAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCGCCAGTATTCTGGCGGGCATGCCTGTTCGAGCGTCATTTCAACCCTCAAGCACAGCTTGGTGTTGGGACTCGCGTTAATTCGCGTTCCCCAAATTGATTGGCGGTCACGTCGAGCTTCCATAGCGTAGTAGTAAAACCCTCGTTACTGGTAATCGTCGCGGCCACGCCGTTAAACCCCAACTTCTGAATGTTGACCTCGGATCAGGTAGGAATACCCGCTGAACTTAAGCATATCAATAAGCGAGAAAAAAAACCAAAACACCACAAA
>11467rpb2
GTAGCGTAGTCGATGTCTCTCTGTGTTGACAGATATACCTTTGCTTCTACTCTTTCACATTTGCGTCGAACCAATACTCCCATCGGACGAGATGGTAAATTGGCCAAGCCTCGACAGCTTCACAACACTCACTGGGGTTTGGTGTGTCCTGCCGAAACACCTGAGGGTCAAGCTTGTGGTCTGGTCAAAAACTTGTCTCTAATGTGTTACGTCAGTGTCGGCTCTCCAGCCGATCCTCTGATTGAATTCATGATCAACAGAGGCATGGAAGTCGTTGAGGAGTACGAGCCGACAAGATACCCCCACGCTACAAAGATTTTCGTCAACGGTAGCTGGGTTGGTGTTCATGCCGACCCCAAGCATCTCGTGAATCAGGTCTTGGACACAAGACGAAAGTCTTACGTGCAGTTCGAAGTATCACTTGTTCGTGATATCCGAGACCGTGAATTCAAGATTTTCTCAGACGCTGGCCGTGTCATGCGACCCGTCTTTACAGTTCATCAGGAGGATGACTATGAGAACAACATCACCAAGGGACAACTAGTGTTGACAAAGGACCATGTCAATAGGCTAGCCCAAGAACAGGCAGAGCCTCCTGCCAACCCAGCGGACAAGTTTGGATGGGATGGCTTGATCCGCGAAGGAGCTGTCGAGTATCTCGATGCTGAGGAAGAAGAGACAGCCATGATTTGCATGACGCCAGAGGATCTCGAACTTTACCGTGAGCAAAAGAATGATGAAGCTACACTCACAGAAGAAGAGAAACGGGCCAAGCAAGAGGCAGAGAAGAGAGAACAAGAGGAGGACCGCAACAAGCGATTGAAGACAAAGGTGAACCCCACAACTCACATGTACACACATTGTGAGATTCACCCTAGTATGATTCTCGGTATCTGTGCCAGTATCATTCCTTTCCCCGATCACAACCAGGTATGTATGTCCTATGACCATGGATAGGCTCAACTAACAGCAATAGTCTCCTCGTATCAGCCTTT
>11467tef1
CCCGTCCCTTTGGGGGGGGGCCCCCCCTACATGTTCTTGATGAAATCACGGTGACCGGGAGCGTCTGAGTGATGTTAGTACGAAGAGAAGTAGAATGAAGCATGAGCGACAACATACCAATGACGGTGACATAGTAGCGAGGAGTCTCGAACTTCCAGAGAGCAATATCGATGGTGATACCACGCTCACGCTCGGCCTTGAGCTTGTCAAGAACCCAGGCGTACTTGAAGGAACCCTTACCGAGCTCAGCGGCTTCCTATTGTTGAATGGTTAGTGACTGCTTGACACGTGACGACGCACTCATTGAGGTTGTGAGAATGGTTAAGAGGGCAAACGCTCCCGTCGCTCAAGTGGCGGGGTAAGTGCCCCACCAAAAAAAATTACAGTCATATTGCAAAATTTTTGGTCTCGAGCGGGGTAGCGGGCACGTTTCGAGTCGTAGGGGAAATCGATGGGCAAAGGACGCGCGATCGAAGGGAAAGTGACTAACCTTCTCGAACTTCTCGATGGTTCGCTTATCGATACCACCGCACTGGTAGATCAAGTGACCGGTCTGTGAAACGATGTCAGTATGTTGACTTTGAGAAATACCCCACCAGGTCTTGGTCGGGATTGACGATGGCAGATAAGCTCATTGTCGAGGAGAGTACTCACAGTGGTCGACTTGCCAGAGTCGACGTGGCCGATGACGACGACGTTAAGGTGAGTCTGCCCCCTCTTTTCCCCAAAAA
cf. Fusarium TUCIM11459
>11459ITS
TGGGACGCGTAGGGTCATTACCGAGTTTACAACTCCCAAACCCCTGTGAACATACCTATACGTTGCCTCGGCGGATCAGCCCGCGCCCCGTAAAACGGGACGGCCCGCCCGAGGACCCCTAAACTCTGTTTTTAGTGGAACTTCTGAGTAAAACAAACAAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCAAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCGCCAGTATTCTGGCGGGCATGCCTGTTCGAGCGTCATTTCAACCCTCAAGCTCAGCTTGGTGTTGGGACTCGCGGTAACCCGCGTTCCCCAAATCGATTGGCGGTCACGTCGAGCTTCCATAGCGTAGTAATCATACACCTCGTTACTGGTAATCGTCGCGGCCACGCCGTTAAACCCCAACTTCTGAATGTTGACCTCGGATCAGGTAGGAATACCCGCTGAACTTAAGCATATCAATAAGCGGAGAAAAAAAAACCCCCCCCCCAAAAAGCACG
>11459rpb2
CGGCTACCTCTGTCTCTAGTGTTGACAGATATACATTTGCTTCCACTCTTTCACATTTGCGACGTACCAACACACCTATTGGTCGTGATGGTAAGCTTGCGAAGCCTCGGCAGCTGCACAACACTCACTGGGGCTTGGTGTGCCCTGCCGAGACACCAGAAGGTCAGGCTTGCGGTCTGGTCAAGAACTTGTCGCTGATGTGCTACGTGAGTGTGGGTTCGCCAGCTGAGCCTTTGATCGAGTTCATGATCAACAGAGGTATGGAAGTTGTTGAAGAATACGAACCAACAAGATATCCTCACGCAACCAAGGTTTTCGTCAACGGAAGTTGGGTCGGCGTCCACCCTGATCCCAGGCACCTGGTGAACTCCGTCCTGGACACACGACGAAAGTCTTACGTCCAGTTCGAGGTTTCCCTTGTTCGTGACATCCGTGACCGTGAATTCAAGATTTTCTCTGATGCAGGCCGAGTCATGAGACCAGTCTTCACAGTGCAACAAGAGGATGATTACGAAACCGGCATCAATAAGGGACAGCTAGTATTGACAAAGGAGCTCGTGAACAAGATTGCCCAGGAGCAGGCAGAGCCACCTGCTGATCCATCGGAGAAGATTGGATGGGAGGGCCTCATCCGCTCTGGAGCTGTGGAGTATCTCGACGCCGAGGAAGAAGAAACCGCAATGATTTGCATGACGCCTGAAGATCTCGAAATCTATCGTGAACAGAAGCAGGACGAGGTTAATCTTACAGAGGAAGAGAAGCGGGCCAAGCAAGAAGCAGAGAAGAGGGAGCAAGAGGAAGAACGCAACAAGCGTCTGAAGACGAAGGTCAACCCCACAACTCACATGTACACACATTGTGAGATTCATCCCAGTATGATTCTGGGTATTTGTGCCAGTATTATTCCCTTCCCCGATCACAACCAGGTATGTATTCCCACCCCCAATCAAGTTGAGCCTATCTAACGACTACAGTCTCCTCGTACACAGATCCCC
>11459tef1
CCCTTTGGGGGGGGGACCCCCCCCCGTAGTTCTTGATGAAATCACGGTGACCGGGAGCGTCTGAAGTACATGTTAGCATGAGAAGGTAATGAGTGCAAGTGATGACAACATACCAATGACGGTGACATAGTAGCGAGGAGTCTCGAACTTCCAGAGGGCGATATCGATGGTGATACCACGCTCACGCTCGGCCTTGAGCTTGTCAAGGACCCAGGCGTACTTGAAGGAACCCTTACCGAGCTCGGCGGCTTCCTATTGTCGGGTGATTAGTGACTGATTGACACGTGATGCGCAAGACATGAGTTTGTGGGAAGAGGGTAAACGCCTGTCACTCGAGTAGCGGGGTTAAAACCCCACCAAAAAAAAATTACGGTTGAACCGCAAAATTCTGTACTCGAGCGGGGTAACAGGCGCATATTCAATCGTCGTAACTGATTCGACTGATGGATCGGTGGGTAGAGGGCGTGCGATCGAGGGAAATGGAAACCAACCTTCTCGAACTTCTCGATGGTTCGCTTGTCGATACCACCGCACTGGTAGATCAAGTGACCGGTCTACGCAATCTTGTCAGCAAATATTCAAGTTGAAATTACCCCGCCACATATGGCGGGGTTGATGACTGCTGATAAGCAGGTCATCGTGGGTAGTACTCACAGTGGTCGACTTGCCAGAGTCGACGTGGCCGATGACGACGACGTTAAGGTGAGCTCCCCCCTTACCCCCCCAAAAAAAAAACCGCCG
cf. Fusarium TUCIM11376
>11376ITS
GTTTGTGACGCGAGGGATCATTACCGAGTTTACAACTCCCAAACCCCTGTGAACATACCTATACGTTGCCTCGGCGGATCAGCCCGCGCCCCGTAAAACGGGACGGCCCGCCCGAGGACCCTAAACTCTGTTTTTAGTGGAACTTCTGAGTATAACAAACAAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCAAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCGCCAGTATTCTGGCGGGCATGCCTGTTCGAGCGTCATTTCAACCCTCAAGCTCAGCTTGGTGTTGGGACTCGCGGTAACCCGCGTTCCCCAAATCGATTGGCGGTCACGTCGAGCTTCCATAGCGTAGTAATCATACACCTCGTTACTGGTAATCGTCGCGGCCACGCCGTAAAACCCCAACTTCTGAATGTTGACCTCGGATCAGGTAGGAATACCCGCTGAACTTAAGCATATCAATAAGCGGAGGAAAAAAAAACCCACCCAAACCTGG
>11376rpb2
ATATACATTTGCTTCCACTCTTTCACATTTGCGACGTACCAACACGCCTATTGGTCGTGATGGTAAACTTGCGAAGCCTCGGCAGCTGCACAACACTCACTGGGGCTTGGTTTGCCCTGCCGAGACACCAGAAGGTCAGGCTTGTGGTCTGGTCAAGAACTTGTCGCTGATGTGCTACGTGAGTGTGGGTTCACCAGCCGAGCCTTTGATCGAGTTCATGATCAACAGAGGTATGGAAGTTGTTGAAGAATACGAACCAACAAGATATCCTCACGCAACCAAGGTTTTCGTCAACGGAAGTTGGGTCGGCGTCCACCCTGATCCCAGGCACCTGGTGAACTCCGTTCTGGACACACGACGAAAATCTTACGTCCAGTTCGAAGTTTCCCTTGTCCGTGACATCCGTGACCGTGAATTCAAAATTTTCTCTGATGCAGGTCGAGTCATGAGACCAGTCTTCACAGTGCAACAAGAGGATGATTACGAAACCGGCATTAATAAGGGACAGCTAGTATTGACAAAGGAGCTCGTGAACAAGATTGCACAGGAGCAAGCAGAGCCACCTGCTGATCCATCGGAGAAGATTGGATGGGAGGGCCTGATCCGCTCTGGAGCTGTGGAGTATCTCGACGCCGAGGAAGAAGAAACCGCAATGATTTGCATGACGCCTGAAGATCTCGAGATCTATCGTGAACAGAAGCAGGACGAGGTTAATCTTACAGAGGAAGAGAAGCGGGCCAAGCAAGAAGCTGAGAAGAGGGAGCAAGAGGAAGAACGCAACAAGCGTCTGAAGACGAAGGTTAACCCCACAACTCACATGTACACACACTGTGAGATTCATCCCAGTATGATTCTGGGTATTTGTGCCAGTATCATTCCCTTCCCCGATCACA
>11376tef1
TCACCTTAACGTCGTCGTCATCGGCCACGTCGACTCTTGCAAGTCGGACCACTGTGAGTGCTACCCTCAATGACCTGTTCATCAACAGTCATTAATTCCGCCATATGGAGCGGGGTCATTTCTACATCTGCTGACAATATTGCATAGACCGGTCACTTGATCTACCAGTGCGGTGGTATCGACAAGCGAACCATCGAGAAGTTCGAGAAGGTTGGTTTCCATTTTCCTCGATCGCGCGCCCTCTGCCCACCGATCCATCACTCGAATCAGTCTCACGACGACTGAATATGCGCCTGTTACCCCGCTCGAGTACAAAAATTTGCGGTTCAATCGTATTTTTTTTTTGGGTGGGGCCTATACCCCGCTACTCGAGTGACAGGCGTTTGCCCTCCCACAATCATCTTGCGCATCACGTGTCAATCAGTCACTAACCACCTGACAATAGGAAGCCGCCGAGCTCGGTAAGGGTTCCTTCAAGTACGCTTGGGTTCTTGACAAGCTCAAGGCCGAGCGTGAGCGTGGTATCACCATCGATATCGCCCTCTGGAAGTTCGAGACTCCTCGTTACTATGTCACCGTCATTGGTATGTTGTTATCACTTACACTCATTGCCTTCTCATGCTAACATGTGTTCCAGACGCTCCCGGTCACCGTGATTTCATCAAGACTG
cf. Fusarium TUCIM11386
>11386ITS
TGGTACGCGTAGGGACATTACCGAGTTTACAACTCCCAAACCCCTGTGAACATACCACTTGTTGCCTCGGCGGATCAGCCCGCTCCCTGTAAAACGGGACGGCCCGCCAGAGGACCCTAAACTCTGTTTCTATATGTAACTTCTGAGTAAAACCATAAATAAATCAAAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCAAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCGCCAGTATTCTGGCGGGCATGCCTGTTCGAGCGTCATTTCAACCCTCAAGCTCAGCTTGGTGTTGGGAACCGCGTTAACCCGCGCTCCCCAAATTGATTGGCGGTCACGTCGAGCTTCCATAGCGTAGTAGTAAAACCCTCGTTACTGGTAATCGTCGCGGCCACGCCGTTAAACCCCAACTTCTGAATGTTGACCTCGGATCAGGTAGGAATACCCGCTGAACTTAAGCATATCAATAAGCGAGAAAAAAAAACAAAACCCAAAAAAAAAAAG
>11386rpb2
GGGAGTGAGCTGAGTCTCTACGTGTGACAGATATACTTTTGCTTCTACTCTTTCACATTTGCGTCGAACCAATACTCCCATTGGACGTGATGGTAAATTGGCCAAGCCTCGTCAGCTTCACAATACTCACTGGGGTCTGGTGTGTCCCGCCGAAACGCCTGAGGGTCAAGCTTGTGGTCTGGTCAAGAACTTGTCTCTGATGTGTTATGTCAGTGTCGGCTCTCCAGCCGAACCTCTCATCGATTTCATGATCAACAGAGGTATGGAAGTCGTTGAGGAGTACGAGCCGACAAGATATCCCCATGCTACAAAGATTTTCGTCAACGGTAGCTGGGTTGGTGTTCACGCCGACCCCAAGCATCTCGTGAATTCGGTTTTGGACACAAGACGAAAGTCGTACGTTCAGTTCGAAGTATCACTTGTTCGTGATATTCGAGACCGTGAATTCAAGATTTTCTCAGACGCTGGTCGTGTCATGAGACCCGTCTTTACGGTCCACCAGGAGGATGACTATGAGAACAACATCACAAAGGGACAGCTAGTGTTGACAAAGGAACATGTCAATAGGCTAGCCCAAGAGCAGGCAGAGCCACCTGCCAACCCCGCGGACAAGTTTGGATGGGATGGCTTGATTCGCGAAGGAGCTGTCGAGTATCTCGACGCTGAGGAAGAAGAGACAGCCATGATTTGCATGACGCCAGAGGATCTCGAACTTTACCGCGAGCAAAAGAATGATGAGGCTACACTCACGGAAGAAGAGAAACGGGCCAAGGCAGAGGCAGAGAAGAGGGAACAAGAGGAGGACCGCAACAAGCGATTGAAGACAAAGGTCAACCCCACAACTCACATGTACACACATTGTGAGATTCACCCCAGTATGATTCTCGGTATCTGTGCCAGTATCATTCCTTTCCCCGATCACAACCAGGTATGTAGCCCCTTTGATCACAAGAAGCTCAACTAACAGCAATAGTCTCCTCGACACATACTTT
>11386tef1
AAAACTTTGGGGGGGGGCCCCCCCTACAGTTCTTGATGAAATCACGGTGACCGGGAGCGTCTGAATGATGGGTTAGTACACAGATGAGTAGAATGAGGCATGAGCGACAACATACCAATGACGGTGACATAGTAGCGAGGAGTCTCGAACTTCCAGAGAGCAATATCGATGGTGATACCACGCTCACGCTCGGCCTTGAGCTTGTCAAGAACCCAGGCGTACTTGAAGGAACCCTTACCGAGCTCAGCGGCTTCCTATTATTGAATGGTTAGTAACCGCTTGACACGTGAGGATGCGCTCATGGTGGGAATGGGAAGAGGGCAAACGCCGTCCGAGTGGCGGGGTAAATGCCCCACCAAAAAAAATTACGGCCATATCGCAAAATTTTTGGTCTCGAGCGGGGTAGCGGGCACGTTTCGAGTCGTAGGAGAAATCGATGGGCAAAGGACGCGCGATCGAAGGGAAAGTGACTAACCTTCTCGAACTTCTCGATGGTTCGCTTGTCGATACCACCGCACTGGTAGATCAAGTGACCGGTCTGTGAAATGATGTCAGCATATTGTCTATCAGAGACACCCGCCAGGTCTTAGTCGGGATAGCTCATCGTCGAGGAAAATACTCACAGTGGTCGACTTGCCAGAGTCGACGTGGCCGATGACGACGACGTTAAGGTGAGCTGCCCCCTCTAAAACACAAAAAAGGAGTGAA
cf. Fusarium s.l. TUCIM11388
>11388ITS
GGAAGCGGAGGACATTACCGAGTTATACAACTCATCAACCCTGTGAACATACCTATAACGTTGCCTCGGCGGGAACAGACGGCCCCGTAACACGGGCCGCCCCCGCCAGAGGACCCCCTAACTCTGTTTCTATAATGTTTCTTCTGAGTAAACAAGCAAATAAATTAAAACTTTCAACAACGGATCTCTTGGCTCTGGCATCGATGAAGAACGCAGCGAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCGCCAGTATTCTGGCGGGCATGCCTGTTCGAGCGTCATTACAACCCTCAGGCCCCCGGGCCTGGCGTTGGGGATCGGCGGAAGCCCCCTGCGGGCACAACGCCGTCCCCCAAATACAGTGGCGGTCCCGCCGCAGCTTCCATTGCGTAGTAGCTAACACCTCGCAACTGGAGAGCGGCGCGGCCACGCCGTAAAACACCCAACTTCTGAATGTTGACCTCGAATCAGGTAGGAATACCCGCTGAACTTAAGCATATCAATAAGCGGAGGAAAGAAAAACCAACCAGGGTGGCTTTCC
>11388rpb2
GGGGGAATGTGCTTGGTGTCTCTAGGTGTTGACAGATACACCTTTGCATCCACTCTTTCACATTTGCGACGAACCAACACCCCTATTGGACGAGATGGAAAGCTCGCCAAGCCCCGTCAGCTACACAACACCCATTGGGGTCTGGTCTGTCCAGCCGAGACGCCTGAGGGTCAGGCTTGTGGTCTGGTCAAGAACTTGTCCCTGATGTGCTACGTCAGTGTCGGCTCTCCCTCTGAACCTCTGATTGAGTTCATGATCAACCGAGGTATGGAAGTCGTGGAAGAGTACGAGCCCCTGAGATACCCGCATGCTACCAAGATCTTTGTCAATGGTGTCTGGTGCGGTGTTCACTCAGACCCCAAGCATCTCGTCCAGCCAGGTTCTGGACACACGACGAAAGTCGTACCTGCAGTATGAGGTGTCGCTTGTTCGTGACATTCGAGATCGAGAGTTCAAGGTCTTCTCCGACGCTGGCCGAGTCATGAGGCCGGTCTTTACGGTCCAGCAGGAGGATGACCACGAGTCTGGTATTGCCAAGGGAGCTTTGGTTCTGACCAAGGACCTTGTCAACAAGCTTGCTAAGGAGCAGGCGGAGCCACCAGAGGACCCATCAATGAAGATTGGATGGGAGGGTCTGATCCGAGCCGGAACCATTGAGTACCTCGATGCTGAGGAAGAGGAGACGGCTATGATTTGCATGACTCCTGAGGATCTTGATCTCTATCGCATGCAAAAGGCTGGTTACGTCGTAGATGACGATAACACGGACGACCCCAACAGGAGATTGAAGACCAAGACGAACCCCACAACTCACATGTACACTCATTGTGAGATTCACCCCAGTATGATTCTTGGCATTTGTGCCAGTATCATTCCCTTCCCCGATCACAACCAGGTATGTGCCCATGATTCAACGTGATGCCAGCGCGGCTAACAATATGTAGTCACCCCGTACACCTCACTCCC
>11388tef1
AGTTTTTTGGAGGTTTCCCCCGGATCATGTTCTTGATGAAGTCACGGTGGCCGGGGGCGTCTGTTGATTGTTAGTGGTGAGACATGTGTGAGAGAGGTGACAGCAACATACCAATGACGGTGACATAGTAGCGGGGAGTCTCGAACTTCCAGAGGGCAATGTCGATGGTGATACCACGCTCACGCTCGGCCTTGAGCTTGTCAAGGACCCAGGCGTACTTGAAGGAACCCTTACCGAGCTCAGCGGCTTCCTATTGTTGGACCGGTTAGCGTCTGTTGTGAACCACGTGATGATGCGCGCCAAGAGGGTTTGGTGTTTTTTGTGTGCAGGGATCAGGGCTTTGTCCAACGTCGCCCGAGTGGCGGGGTAAATGCCCCACCAAAAAAATTACGGTCGGACCGCAAAAATTTTGGGACTCGGGAGAAGCGGGCAGAGCGTATCGCGAGGGAGGGAATTCGATGTGGAATAGCAAGGCGCGATCGGGGGAGATGTCACCAACCTTCTCGAACTTCTCGATGGTTCGCTTGTCGATACCACCGCACTGGTAGATCAAGTGACCGGTCTGTAGATGATTGTCAGCATGAAGTGACTGATGAATACCCCGCCCGAGATGCCAGGCGGGGTTCCACGACCCGAGATAAGCAGATCGCGATGAGGGTTTGACTTACGGTGGTCGACTTGCCAGAGTCGACGTGGCCGATGACGACGACGTTGAGGTAGCTCCCCCCCCTCCCCCCAAAAAAAAAGTTAGG
cf. Fusarium s.l. TUCIM11430
>11430ITS
AGCGTAGGGACATTACCGAGTTATACAACTCATCAACCCTGTGAACATACCTAAAACGTTGCTTCGGCGGGAACAGACGGCCCCGTAACACGGGCCGCCCCCGCCAGAGGACCCCCTAACTCTGTTTCTATTATGTTTCTTCTGAGTAAAACAAGCAAATAAATTAAAACTTTCAACAACGGATCTCTTGGCTCTGGCATCGATGAAGAACGCAGCGAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCGCCAGTATTCTGGCGGGCATGCCTGTTCGAGCGTCATTACAACCCTCAGGCCCCCGGGCCTGGCGTTGGGGATCGGCGAGGCGCCCCCTGCGGGCACACGCCGTCCCCCAAATACAGTGGCGGTCCCGCCGCAGCTTCCATTGCGTAGTAGCTAACACCTCGCAACTGGAGAGCGGCGCGGCCACGCCGTAAAACCCCCAACTTCTGAATGTTGACCTCGAATCAGGTAGGAATACCCGCTGAACTTAAGCATATCAATAAGCGGAGGAAAGAAAAACCAACCAATTTATTTAT
>11430rpb2
TGCTGTGTCTCTAGGTGTGACAGATACACCTTTGCATCCACTCTTTCACATTTGCGACGAACCAACACCCCTATTGGACGAGACGGAAAGCTCGCCAAGCCCCGTCAGCTACACAACACCCATTGGGGTCTGGTCTGTCCAGCCGAGACGCCTGAGGGTCAGGCTTGTGGTCTGGTCAAGAACTTGTCCCTGATGTGCTACGTCAGTGTCGGCTCTCCCTCTGAACCTCTGATTGAGTTCATGATCAACCGAGGTATGGAAGTCGTGGAAGAGTACGAGCCCCTGAGATACCCGCATGCTACCAAGATCTTTGTCAATGGTGTCTGGTGCGGTGTTCACTCAGACCCCAAGCATCTCGTCAGCCAGGTTCTGGACACACGACGAAAGTCGTACCTGCAGTATGAGGTGTCGCTTGTTCGTGACATTCGAGATCGAGAGTTCAAGGTCTTCTCCGACGCTGGCCGAGTCATGAGGCCGGTCTTTACGGTTCAGCAGGAGGATGACCACGAGTCTGGTATTGCCAAGGGAGCCTTGGTTCTGACCAAGGACCTTGTCAACAAGCTTGCTAAGGAGCAGGCGGAGCCACCAGAGGACCCATCAATGAAGATTGGATGGGAGGGTCTGATCCGAGCCGGAACCATCGAGTACCTCGATGCTGAGGAAGAGGAGACGGCTATGATTTGCATGACTCCTGAGGATCTTGATCTCTATCGCATGCAAAAGGCTGGTTACGTCGTAGATGACGATAACACGGACGACCCCAACAGGAGATTGAAGACCAAGACGAACCCCACAACTCACATGTACACTCATTGTGAGATTCACCCCAGTATGATTCTTGGCATTTGTGCCAGTATCATTCCCTTCCCCGATCACAACCAGGTATGTGCCCATGATTCAACGTGATGCCAGCGCGGTAACAATATGTAGTCCCCCGTAAACTACATGCGGGGGGGGGAAA
>11430tef1
CCTTTTGGGGGGGGCCCCCCCCCCGCAGTTTTGATGAAATCACGGTGGCCGGGGGCGTCTGTTGATTGTTAGTGGTGAGACATGTGTGAGAGAGGTGACAGCAACATACCAATGACGGTGACATAGTAGCGGGGAGTCTCGAACTTCCAGAGGGCAATGTCGATGGTGATACCACGCTCACGCTCGGCCTTGAGCTTGTCAAGGACCCAGGCGTACTTAAAGGAACCCTTACCGAGCTCAGCGGCTTCCTATTGTTGGACCGGTTAGCGTCTGTTGTGAACCACGTGATGATGCGCGCCAAGAGGGTTTGGTGTTTTTTGTGTGCAGGGATCAGGGCTTTGTCCAACGTCGCCCGAGTGGCGGGGTAAATGCCCCACCAAAAAAATTACGGTCGGACCGCAAAAATTTTGGGACTCGGGAGAAGCGGGCGCAGACCGTATCGCGAGGGAGGGAATTCGATGTGGAATAGCAAGGCGCGATCGGGGGAGATGTCACCAACCTTCTCGAACTTCTCGATGGTTCGCTTGTCGATACCACCGCACTGGTAGATCAAGTGACCGGTCTGTAGATGATTGTCAGCATGAAGTGACTGATGAATACCCCGCCCGAGATACCAGGCGGGGTTCCACGACCCGAGATAAGCAGATCGAGATGAGGGTTTGACTTACGGTGGTCGACTGCCAGAGTCGACGGCCGAGACGACACATGC
cf. Fusarium s.l. TUCIM11465
>11465ITS
TGAGGGTCGCGGAGGACATTACCGAGTTATACAACTCATCAACCCTGTGAACATACCTAAACGTTGCCTCGGCGGGAACAGACGGCCCCGTGAAAACGGGCCGCCCCCGCCAGAGGACCCCCTAACTCTGTTTCTATAATGTTTCTTCTGAGTAAAACAAGCAAATAAATTAAAACTTTCAACAACGGATCTCTTGGCTCTGGCATCGATGAAGAACGCAGCGAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCGCCAGTATTCTGGCGGGCATGCCTGTTCGAGCGTCATTACAACCCTCAGGCCCCCGGGCCTGGCGTTGGGGATCGGCGGAGGCCCTCCGTGGGCACACGCCGTCCCCCAAATACAGTGGCGGTCCCGCCGCAGCTTCCATCGCGTAGTAGCTAACACCTCGCGACTGGAGAGCGGCGCGGCCACGCCGTAAAACCCCCAACTCTTCTGAAGTTGACCTCGAATCAGGTAGGAATACCCGCTGAACTTAAGCATATCAATAAGCGGAGGAAAAAAAAACCCCACCCAATTTTT
>11465rpb2
GATACACCTTTGCATCCACTCTTTCGCATTTGCGACGAACCAACACCCCTATTGGACGAGATGGAAAGCTCGCCAAGCCCCGTCAGCTACACAACACCCATTGGGGTCTGGTCTGTCCAGCCGAGACGCCCGAGGGTCAGGCTTGTGGTCTGGTCAAGAACTTGTCCCTGATGTGTTACGTCAGTGTCGGTTCTCCTTCTGAACCTCTGATCGAGTTCATGATCAACCGAGGCATGGAAGTCGTGGAAGAGTACGAGCCCCTGAGATACCCGCATGCTACCAAGATCTTTGTCAATGGTGTCTGGTGCGGTGTCCACTCGGACCCCAAGCATCTCGTCAGCCAGGTCCTGGACACACGACGAAAGTCGTACCTGCAATACGAGGTGTCGCTTGTCCGTGACATTCGAGATCGAGAGTTCAAGGTCTTCTCCGACGCTGGCCGAGTCATGAGGCCAGTCTTTACGGTTCAGCAGGAGGATGACCACGAGTCTGGTATCGCCAAGGGAGCTTTGGTTCTGACCAAGGACCTTGTCAACAAGCTTGCTAAGGAGCAGGCGGAACCACCAGAGGACCCATCAATGAAGATTGGATGGGAGGGTCTGATCCGAGCCGGAACCATCGAGTACCTCGATGCTGAGGAAGAGGAGACGGCTATGATTTGCATGACTCCTGAGGATCTTGACCTCTATCGCATGCAAAAGGCTGGTTACGTCGTAGATGACGATAACACGGACGACCCCAACAGGAGATTGAAGACTAAGACGAACCCCACAACTCACATGTACACTCATTGTGAGATTCACCCCAGTATGATTCTTGGCATTTGTGCCAGTATCATTCCCTTCCCCGATCACAACCAGGTATGCGCCCATGATTCAACGTGATGCTAGCGCAGCTAACAAT
>11465tef1
TGGAGGTACCCCCAATGATCATGTTCTTGATGAAATCACGGTGGCCGGGGGCGTCTGTTGATTGTTAGTGATGAGACGTGATTGAGAGAGATGAGGGCGACATACCAATGACGGTGACATAGTAGCGGGGAGTCTCGAACTTCCAGAGAGCAATATCGATGGTGATACCACGCTCACGCTCGGCCTTGAGCTTGTCAAGGACCCAGGCGTACTTGAAGGAACCCTTACCGAGCTCAGCGGCTTCCTATTGTTGAACCTGTTAGTGTCTGTTGTGAACCACGTGATGCGCGCCAAGAGGGTTTGGTGTTTTTTGTGTGCAGGGTTCAGGGCTCGTCCAACGTCGCCCGAGTGGCGGGGTAAATGCCCCACCAAAAAAATTACGGTCGAACCGCAAAATTTTTGGGACTCGGAAGAAGCGGGCGCAGAGCGTGTCGCGGAAGAGGGAATTCGACGGGGAATTCGATGTGGAATAGCAAGGCGCGATCGGGGGAGATGTCACCAACCTTCTCGAACTTCTCGATGGTTCGCTTGTCGATACCACCGCACTGGTAGATCAAGTGACCGGTCTGTAAATGTTTGTCAGCATGAAGTGACTGATGAGTACCCCGCCCGAGATATCAGGCGGGGTTCCACGACCCGAGATAAGCAGATCGCGATGAGGGTTTGACTTACGGTGGTCGACTGCCAGAGTCGACGTGCCGATTACGAGAACAATGG
cf. Fusarium s.l. TUCIM11357
>11357ITS
TAGGGGTCCGGGGGGGAAGCGGAGGACATTACCGAGTTATTCACTCATCAACCCTGTGAACATACCTAAACGTTGCTTCGGCGGGAACAGACGGCCCCGTGAAACGGGCCGCCCCCGCCAGAGGACCCCTAACTCTGTTTCTATAATGTTTCTTCTGAGTAAAACAAGCAAATAAATTAAAACTTTCAACAACGGATCTCTTGGCTCTGGCATCGATGAAGAACGCAGCGAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCGCCAGTATTCTGGCGGGCATGCCTGTTCGAGCGTCATTACAACCCTCAGGCCCCCGGGCCTGGCGTTGGGGATCGGCGGAGCCCCCCGTGGGCACACGCCGTCCCCCAAATACAGTGGCGGTCCCGCCGCAGCTTCCATCGCGTAGTAGCTAACACCTCGCGACTGGAGAGCGGCGCGGCCACGCCGTAAAACACCCAACTCTTCTGAAGTTGACCTCGAATCAGGTAGGAATACCCGCTGAACTTAAGCATATCAATAAGCGGAGGAAAAAAAAAACCCAACC
>11357rpb2
GGCAACACTGCTGCTGTCTCTAGGTGTTGACAGATACACCTTTGCATCCACTCTTTCGCATTTGCGACGAACCAACACCCCTATTGGACGAGATGGAAAGCTCGCCAAGCCCCGTCAGCTACACAACACCCATTGGGGCCTGGTCTGTCCAGCCGAGACACCCGAGGGTCAGGCTTGTGGTCTGGTCAAGAACTTGTCCCTGATGTGCTACGTCAGTGTCGGTTCTCCCTCCGAGCCTCTGATTGAGTTCATGATCAACCGAGGTATGGAAGTCGTGGAGGAGTACGAGCCCCTGAGATACCCACATGCCACCAAGATCTTTGTCAATGGTGTCTGGTGTGGTGTCCACTCGGACCCTAAGCATCTCGTCAGCCAGGTTCTGGACACACGACGAAAGTCGTACCTGCAGTATGAGGTGTCGCTTGTTCGTGACATTCGAGATCGAGAGTTCAAGGTCTTCTCCGACGCTGGCCGAGTCATGAGGCCGGTCTTTACGGTCCAGCAGGAGGATGACCACGAGTCTGGTATCGCCAAGGGAGCTTTGGTTCTGACCAAGGACCTTGTCAACAAGCTTGCTAAGGAGCAGGCGGAGCCACCAGAGGACCCATCAATGAAGATTGGATGGGAGGGTCTGATCCGAGCCGGAACTATCGAGTACCTCGATGCTGAGGAAGAGGAGACGGCTATGATTTGCATGACTCCTGAGGATCTTGACCTCTATCGCATGCAAAAGGCTGGTTACGTCGTAGATGACGATAACACGGACGACCCCAACAGGAGATTGAAGACCAAGACGAACCCCACAACTCACATGTACACTCATTGTGAGATTCACCCCAGTATGATTCTTGGCATTTGTGCCAGTATCATTCCCTTCCCCGATCACAACCAGGTATGTGCCCTTGTTTCAACGTGATGCTAGCACAGCTAATAATACGTAGTCACCTCGTACCCCTCCTTTTCG
>11357tef1
CGGCAGTTCTTGATGAAATCACGGTGGCCGGGGGCGTCTGTTGATTGTTAGTGATGAGACGTGAGTGAGAGAGATGACGGCGACATACCAATGACGGTGACATAGTAGCGGGGAGTCTCGAACTTCCAGAGAGCAATATCGATGGTGATACCACGCTCACGCTCGGCCTTGAGCTTGTCAAGGACCCAGGCGTACTTGAAGGAACCCTTACCGAGCTCAGCGGCTTCCTATTGTTGAACCAGTCAGTGTCTGTTGTGAACCACGTGATGCGCGCCAAGAGGGTTTGGTGTTTTTGTGTGCAGGGATCAGGGCTTTGTCCAACGTCGCCCGAGTGGCGGGGTAAATGCCCCACCAAAAAAATTACGGTCGAACCGCAAAATTTTTGGGACTCGGGAGAAGCGGGCGCAGAGCGTGTCGCGGAGGAGGGAATTCGACGGGGAATTCGATGTGGGATAGCAAGGCGCGATCGGGGGAGATGTGACCAACCTTCTCGAACTTCTCGATGGTTCGCTTGTCGATACCACCGCACTGGTAGATCAAGTGACCGGTCTGTAGATGATTGTCAGCATGAAGTGACTGATGAGCACCCCGCCCGAGATTCCAGGCGGGGTTCCACGACCCGAGATAAGCAGATCGCGATGAGGGTTTGACTTACGGTGGTCGACTTGCCAGAGTCGACGTGGCCGATGACGACGACGTTGAGGTGAGCTGTCC
cf. Parasarocladium TUCIM11530
>11530ITS
GGTTTGTGGGGACGCGGAGGGACATTATGAGTTTAACGCTCTTCAACCCCTGTGAACCATACCTATTTTTCCCCGGCGGAACGCCCCGGTGCCTTAGGGGCCGGGTCAGGCGCCCGCCGGGGGCAAACAGACTGTATTTGTACCCCTATACCTCAGAATTAGGCGTAAAGCCGAAATGTAACAACTTTTGACAATGGATCTCTTGGCACTAGCATCGATGAAGAACGCAGCGAAATGCGATAAGTAATGTGAATTGCAGAGCACAGTGAATCATCGAATCTTTGAACGCACATGGCGCCCGCCGGTAATCCGGTGGGCATGCCTGTCCGAGCGTCATTTCAACCCTCAGGACCCCTTCGGGGCGTCACTGGTGTTGGGGATCGAGGGCCGTCCAGGCTCCCGCCCCCTAAATTCATTGGCGGCTTCCGGAGTTGTCTCCCCTGCGTAGTACTGCATCTCGCTTTGGTGACTCCCCCGGGCCGCTGCGGCCCCGCGCCGCGAACCCCCCAAGGTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAAGCATATCAATAAGCGGAGGAAAAAAA
>11530rpb2
CCCCCTGCTGATGTGTCTCACTTTGACAGATACACCTTCGCCTCCACACTGTCGCATTTGCGACGAACCAACACACCCATCGGCAGAGATGGAAAGCTGGCCAAGCCCCGCCAACTGCACAACACACATTGGGGTCTCGTGTGTCCTGCCGAGACGCCCGAGGGACAAGCTTGTGGTCTGGTCAAGAACTTGTCGCTTATGTGCTATGTCAGTGTTGGTTCGCCCTCTGACCCCATCATCGAGTTCATGATCAACCGTGGTATGGAGGTCGTTGAGGAGTACGAGCCCATGCGTTTCCCCAACGCTACCAAGGTCTTCGTTAACGGTACGTGGTGCGGTGTCCACCAGGATCCCAAGCACTTGGTCGACCAGCTTGTCACCACCCGACGAAAGGGTTTCATTTCGGATGAAGTCTCACTCGTTCGCGATATCCGTGATCGTGAGTTCAAGGTATTTTCAGATGCTGGCCGTGTCATGCGTCCAGTCTTCACAGTCGAGCAAAACGATGACTCGGAGAGCGGTGTCGAAAAGGGTGCCATGCTTCTGACCAAGGATATGGTCAACAGACTGGACAAGGAGAGGAACAAGGACTATGCCGAGAACGAGAGACTGGTCTGGAAGGACCTTACAGAGCGAGGTGCGGTCGAGTACCTTGATGCTGAGGAAGAAGAAACGGCCATGATCTGCATGACACCTGAGGATCTTGAGTTGTACCGCTTGCAAAAGGCGGGACATGTCATTCCCCCAGAGGATCCTGGTGATGATCCCAACAAGCGTCTCAAGACAAAGCACAACCCGACTACACACACATACACTCACTGCGAGATTCATCCCAGTATGATTCTGGGCATTTGCGCCAGTATCATTCCCTTCCCCGATCACAACCAGGTAGGTTCTGCTCTGTCACCATCATCATGTGCTGTATGACAAAACCAGTCCCACCGTGCACTAGAT
cf. Penicillium TUCIM11507
>11507ITS
GTAGTACTCGGAGGACATTACCGAGTGAGGGCCCTCTGGGTCCAACCTCCCACCCGTGTTTATCGTACCTTGTTGCTTCGGCGGGCCCGCCTCACGGCCGCCGGGGGGCATCCGCCCCCGGGCCCGCGCCCGCCGAAGACACCAATGAACTCTTGTCTGAAGATTGCAGTCTGAGCAGATTAGCTAAATCAGTTAAAACTTTCAACAACGGATCTCTTGGTTCCGGCATCGATGAAGAACGCAGCGAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAGTCTTTGAACGCACATTGCGCCCCCTGGTATTCCGGGGGGCATGCCTGTCCGAGCGTCATTGCTGCCCTCAAGCACGGCTTGTGTGTTGGGCTTCGCCCCCCGGCTCCCGGGGGGCGGGCCCGAAAGGCAGCGGCGGCACCGCGTCCGGTCCTCGAGCGTATGGGGCTTTGTCACCCGCTCTGTAGGCCCGGCCGGCGCCCGCCGGCGACCCCAAATCAATCTATCCAGGTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAAGCATATCAATAAGCGAGGAAGAAAAAACCCCCCCCCCAACCCTTGG
>11507rpb2
GAGCTCGGCTGATGTGTCAGGTGCTGAGTCGATACACCTTTGCTTCCAGTTTGTCCCATCTACGTCGTACCAACACACCCATTGGTCGAGATGGTAAGATCGCCAAACCGCGTCAACTTCACAACACCCACTGGGGTCTCGTCTGTCCTGCCGAGACTCCTGAAGGTCAAGCTTGTGGTCTCGTCAAGAACCTGGCACTCATGTGCTATATCACCGTTGGTACACCTAGTGAGCCCATCGTTGATTTCATGATTCAGCGGAACATGGAAGTTCTTGAAGAGTTCGAACCCCAGGTCACGCCTAATGCCACCAAGGTGTTCGTCAACGGCGTCTGGGTTGGTATCCACCGTGACCCCACACATCTAGTCAACACGATGCAGTCTCTTCGTCGGCGCAACATGATCTCCCACGAGGTCAGCTTGATTCGCGACATCCGTGACCGAGAGTTCAAGATCTTCACAGATGCTGGACGTGTGTGCCGACCCCTGTTCGTCGTTGACAACGACCCCAAGAGTGAGAACGCTGGATCCTTGGTCCTCAATAAGGAGCACATTCGCCTGCTGGAGCTGGACAAGGATCTTCAACGAGAACTGCCCCCAGAGCAGCTCCGGGAGCAAGGCTTCGGATGGGAGGGTTTGGTCAGATCGGGTGTTGTTGAATACGTGGATGCCGAGGAAGAAGAGACGATCATGATCGTCATGACCCCTGAAGACTTGGAGATTTCCAAGCACCTCCAAGCCGGTTACTCTCTGCCAGAGGATGACAAGAACGACCCCAACAAACGAGTACGCACCGTGACCCAACGTGCGCATACCTGGACGCATTGCGAGATTCATCCCAGTATGATTCTGGGTGTGTGTGCCAGTATCATTCCTTCCCCGACCACAACCAGTCTCCTCGTACCCCCGACCC
>11507cal
GGGGGGGAAGGTGCTCTCGACGCGTCATTGACAATTCCGATCGGAAAAAAACACAGTTGACTGACCAAGGCCGGTTTTCCCGTGAACAGGACAAGGATGGCGATGGTGAGTGCAGTCTTCTCTAGTAGCTCGGGCGCTCTATCTCAGGAAGCATGCTTTGGGCCATTGGATATTGTGAGACTTCTATAACAGTGTTGCTAATCCATATACCTTCATTAGGACAAATCACCACCAAGGAGCTCGGCACTGTCATGCGCTCGCTCGGCCAGAACCCCTCCGAGTCCGAGCTGCAGGATATGATCAACGAGGTTGATGCCGACAACAACGGCACCATCGACTTCCCCGGTACGCCATTCCTGCCTTGGGCTATTATTTGCCGCTATTTTCCCTGTAGCCTGAGACTAATTGCTTTTCTGCGCCCACAGAGTTCCTCACCATGATGGCCCGTAAGATGAAGGACACCGACTCAGAGGAGGAGATCCGGGAAGCGTTCAAGGTGTTCGACCGTGACAACAACGGCTTCATCTCCGCCGCCGAGCTGCGCCACGTTATCCCC
>11507tub
GGTGGTGACGGCCGCCAAATCTCCCCAGCATAACAACGCAACACCCCAATTGCGAGTCTAATGAATCGAATTATGCTGACTCGAACTACAGGCAGACCATTGCTGGTGAGCACGGCCTTGACGGCGATGGCCAGTAAGTTTCTTCAATAACCTCCCCTTCGACAATCCCACTCGACGCGGAATGGCGGTCTGATATTTTTGGCTAGCTACAACGGTACCTCCGACCTCCAGCTGGAGCGCCTGAACGTCTACTTCACCCACGTAAGTTGCGTGTCCATCGAAGAGATACTCGGAGTCATGTCTAATCAATGCATCTTCTGTTGTTCTAGGCCAGCGGTGACAAGTATGTTCCCCGTGCTGTTCTCGTCGATCTTGAGCCCGGTACCATGGACGCTGTCCGTGCCGGTCCCTTTGGCAAGCTCTTCCGTCCCGACAACTTCGTCTTCGGTCAGTCCGGTGCCGGTAACAACTGGGCCAAGGGTCACCA
cf. Penicillium TUCIM11508
>11508ITS
TAAAGTGACTGCGGAGGACATTACCGAGTGAGGGCCCTCTGGGTCCAACCTCCCACCCGTGTTTATCGTACCTTGTTGCTTCGGCGGGCCCGCCTCACGGCCGCCGGGGGGCATCCGCCCCCGGGCCCGCGCCCGCCGAAGACACACAAACGAACTCTTGTCTGAAGATTGCAGTCTGAGTACTTGACTAAATCAGTTAAAACTTTCAACAACGGATCTCTTGGTTCCGGCATCGATGAAGAACGCAGCGAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAGTCTTTGAACGCACATTGCGCCCCCTGGTATTCCGGGGGGCATGCCTGTCCGAGCGTCATTGCTGCCCTCAAGCACGGCTTGTGTGTTGGGCTCTCGCCCCCCGCTTCCGGGGGGCGGGCCCGAAAGGCAGCGGCGGCACCGCGTCCGGTCCTCGAGCGTATGGGGCTTCGTCACCCGCTCTGTAGGCCCGGCCGGCGCCCGCCGGCGAACACCATCAATCTTAACCAGGTTGACCTCGGATCAGGTAGGGATACCCGCTGAACTTAAGCATATCAATAAGCGGAGGAAGAAAAAACCCAACCAAAACTTTTTA
>11508rpb2
GGGGATTGCTGATGTCTCAGTGCTTAGTCGTTACACATTTGCTTCTAGTTTGTCTCATCTTCGTCGTACCAACACACCTATTGGCCGAGATGGTAAGATTGCGAAGCCTCGTCAGCTACATAATACTCACTGGGGTCTTGTCTGTCCGGCCGAGACACCAGAAGGTCAAGCTTGTGGTCTTGTCAAGAACTTGGCTCTGATGTGTTACATCACGGTCGGAACGCCCAGCGAGCCTATCGTGGACTTCATGATTCAACGGAACATGGAGGTTCTTGAAGAGTTCGAGCCACAGGTTACTCCCAACGCTACGAAGGTCTTTGTTAATGGTGTCTGGGTCGGTATTCACCGTGATCCGACACACCTGGTCAACACGATGCAGTCCTTGCGTCGACGCAACATGATCTCTCATGAAGTCAGCTTGATTCGTGACATTCGTGACCGAGAATTTAAAATTTTCACGGATGCTGGACGTGTCTGCCGTCCCTTGTTTGTTGTGGACAATGACCCGAAGAGTGAAAATGCAGGATCTCTGGTCCTGAATAAGGAACATATTCGTTTGCTTGAGTTAGACAAGGATCTTCAACGTGAGCTGCCACCAGAGCAACTCCGCGAGCAGGGTTTCGGATGGGACGGCTTGGTCAGGTCAGGAGTCGTGGAATACGTCGACGCTGAAGAGGAAGAGACGATTATGATTGTCATGACCCCCGAGGATCTTGAGATCTCCAAACATCTTCAGGCAGGATATTCCCTCCCCGAAGATGACAAGAACGATCTCAACAAGCGTGTCCGCACTGTCACTCAGCGCGCCCACACTTGGACTCACTGCGAGATTCATCCCAGTATGATTCTTGGTGTCTGTGCCAGTATCATTCCTTCCCCGATCACAACCAGTCGCCCCGTAACCCTTACC
>11508cal
GGTTTGATGTCCAATGACGAGCATTGGGACTTCCCATGTGAAAGAGGAAATCCAACAACAAACATGGTAAACTAATGGGGCTGTTTTTTTCCCGCGTGTAGGACAAGGATGGCGATGGTGAGATTCTTCCTCTCTCCCCTCCCCCCACCGATTCTTTGTGCATGGTTGCAAGGAATCTTGACCGGAATGATGAATGCTCGGTGCTTCCCGTAGCACAGTGGTCGAATGGGTATAGCTGACTCGAGGGTTGCAATCAGGCCAAATCACCACCAAGGAGCTCGGTACTGTCATGCGCTCGCTTGGCCAGAACCCCTCCGAGTCTGAGCTGCAGGACATGATCAACGAGGTCGATGCCGACAACAACGGCACCATTGACTTCCCTGGTACGCTTTTCCCTAGTACCGTATCTTGCAACCCGGTGTCACCTTTTTTTTTGGGTGCCCTGAGCGCAGTGTAATTAATTGTGGCCCGCACCGACAGAGTTCCTCACGATGATGGCCCGCAAGATGAAGGACACCGATTCCGAGGAGGAGATCCGTGGAGGCCTTCAAGGTCTTTGACCGTGATAACAACGGCTTCATTTCCGCCGCTGAGCTGCGCCACGTTGCCCCCCCTTTTTAGGGGGCTCCCCCCCCCACCCG
>11508tub
TTATGACCCTTGGCCCAGTTGTTACCAGCACCAGACTGGCCAAAGACGAAGTTGTCGGGGCGGAAGAGCTTGCCAAAGGGACCGGCACGGACAGCGTCCATGGTACCAGGCTCCAGGTCGACGAGAACGGCACGGGGAACATACTTGTCACCGCTAGCCTGGAAAATCGAACGTTTCAATGATCAGGTTTGGGAGATTTCTTTGATTGGGAAGACGTTTTTCACGACTTACGTGGGTGAAGTAGACGTTCATGCGCTCCAGCTGGAGGTCGGAGGAACCATTGTAGCTAAACCGAAAAATATCAGACCGCCATTCTCTGTCGTCAGGATCCCATCGTGGGGGGGGGGCAAGCTGTTGACTTACTGGCCATCGCCGTCAAGGCCGTGCTCACCAGCAATTGTTTGCCTGAATATCGAGTCAGTTTGGTTCTCATATTAGATCGAATCACGACACAAGGAATTGTCGGTGTTGTGCTCGGGATATCCCAG
cf. Plectosphaerella TUCIM11531
>11531ITS
CGGAGGACATTCTGAGTCTACACTCTCTACCCTTTGTGAACTATTATACCTGTTGCTTCGGCGGCGCCCGCGAGGGCGCCCGCCGGTCTCATCAGAATCTCTGTTTTCGAACCCGACGATACTTCTGAGTGTTCTTAGCGAACTGTCAAAACTTTTAACAACGGATCTCTTGGCTCCAGCATCGATGAAGAACGCAGCGAAACGCGATATGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATGGCGCCTTCCAGTATCCTGGGAGGCATGCCTGTCCGAGCGTCGTTTCAACCCTCGAGCCCCCCGTGGCCCGGCGCTGGGGATCTGCCACGGCAGGCCCCTAAAACCAGTGGCGGACCCGCTGGGCCCTCTCCTTTGCGCAGTAGCATCAGCCTCGCATCGGGAGCCCTCGGCGTCCTGCCTCTAAACCCCCCACAAGCCCGCTCCGGCGGCACCAAGGTTGACCTCGGATCAGGTAGGAATACCCGCTGAACTTAAGCATATCAATAAGCGGAGGAAAAGAAAAACCCAACCAGAGGGAG
>11531rpb2
GCCGACGGCTGATGTCTCTAGGTGCTCACAGATACACCTTTGCCTCGACGCTGTCGCATTTGCGACGCACCAACACCCCCATCGGTCGTGACGGAAAGCTCGCCAAGCCCCGCCAGCTTCACAACACGCATTGGGGTCTTGTGTGTCCCGCCGAGACGCCCGAGGGTCAGGCTTGCGGTCTCGTCAAGAACCTCTCCCTAATGTGCTACGTCAGCGTGGGTACGCCAGCCGAGCCCATCATTGACTACATGATCCAGCGAGGCATGGAAGTTCTCGAGGAGTACGAGCCGCTCCGATACCCCAACGCCACAAAGGTCTTCCTGAACGGCGTGTGGGTGGGTGTGCACCAAGACCCGAGGACTCTCGTCCCGGACGTCCAGGGCACGCGCCGTCGCAACGTTATTTCTCACGAGGTGTCGCTCGTCCGCGATATCCGCGATCGTGAGTTCAAGATATTTTCCGATGCCGGCCGTGTGATGAGACCAGTCTTTGTTGTTGAGCAGAGCACCACGGGCACCGTCGAGCAGGGCCACCTCGTCCTGCAGAAGGCCCTGGTCAACGAGATGCGTGAGCAGCAGGAGGTGCCCCCGGACAACCCCGACGACAAGATCACCTGGCAGACGCTGGCCCACAGCGGTGTCATCGAGTACCTTGACGCCGAGGAGGAGGAGACGTCCATGATCTGCATGACGCCGGAAGACCTGGAGCTGTTCCGCAACCAGAAGGCCGGGTACGAGTACCCCGACGACGTCGCAGAAGGCCCCAACAGGCGTCTGCGGACCAAGATTGCCAAGACGACGCACATGTACACGCATTGCGAGATTCACCCGAGTATGCTTCTCGGTATTTGCGCAAGCATCATTCCCTTCCCCGACCACAACCAAGTAAGTATGCCCTTTACAGCCTTGAGATCCCATCTGCTAACACGCCGCAGTCTCCCCGTACGCCTCCCC
>11531tub
GAGGGGGACGTCCCGACGACGATGCATCTCGACGTGATCGATTGAATACAGAATACTGATTCCCGCCACAGGCAGAACATCTCTGGCGAGCACGGTCTCGACGCCAATGGCGTGTATGTTCACAATTCCCTGCCGGGAACCTCCACGACAGCCTTGCTGACACGAGGAAAACAGCTACGCCGGCAGCTCTGAGCTCCAGCTTGAGCGCATGAACGTCTACTTCAACGAGGTCAGCGATACACCTTCACTTCAATTCGATTCTCATTGCTAACTCGGTGACAACAGGCCTCCAGCAACAAGTACGTTCCCCGTGCCGTCCTCGTCGATCTCGAGCCCGGTACCATGGATGCCATCCGTGCCGGTCCCTTCGGCCAGCTCTTCCGCCCCGACAACTTCGTCTTCGGCCAGTCCGGTGCCGGAAACAACTGGGCCAAGGGTCATCA
cf. Plectosphaerella TUCIM11532
>11532ITS
AGGTACGCGTAGGGTCATTACTGAGTACTACACTCTCTACCCTTTGTGAACTATTATACCTGTTGCTTCGGCGGCGCCCGCAAGGGTGCCCGCCGGTCTCATCAGAATCTCTGTTTTCGAACCCGACGATACTTCTGAGTGTTCTTAGCGAACTGTCAAAACTTTTAACAACGGATCTCTTGGCTCCAGCATCGATGAAGAACGCAGCGAAACGCGATATGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATGGCGCCTTCCAGTATCCTGGGAGGCATGCCTGTCCGAGCGTCGTTTCAACCCTCGAGCCCCTGTGGCCCGGCGTTGGGGATCTGCCACGGCAGGCCCCTAAAACCAGTGGCGGACCCGAAGGGCCCTCTCCTTTGCGCAGTAGCATTCGCCTCGCATTGGGAGTCCTCGGCGTCCTGCCTCTAAACCCCCCACAAGTCCGCTTCGGCGGCACCAAGGTTGACCTCGGATCAGGTAGGAATACCCGCTGAACTTAAGCATATCAATAAGCGAGGAAGAAAACACCCCCCCCCAAAGGGTGCCG
>11532rpb2
GGCGGGAACGATGATGTCTCAGGTGCTCACAGATACACCTTTGCCTCGACGCTGTCGCATTTGCGCCGCACCAACACGCCCATCGGTCGTGACGGAAAGCTCGCCAAGCCTCGTCAGCTTCACAACACGCATTGGGGTCTCGTTTGTCCCGCCGAGACGCCCGAGGGTCAGGCTTGTGGTCTCGTCAAGAACCTCTCCCTCATGTGCTACGTCAGTGTGGGCACGCCCGCTGAGCCCATCATCGACTATATGATTCAGCGTGGCATGGAAGTGCTCGAGGAGTACGAGCCGCTCCGATACCCCAACGCCACAAAGGTCTTCCTCAACGGCGTGTGGGTGGGTGTGCACCAAGACCCGAGAACTCTCGTCCCGGACGTCCAGGGCACGCGCCGTCGCAACGTTATTTCTCACGAGGTGTCGCTTGTCCGCGATATCCGCGATCGTGAGTTCAAGATATTTTCCGATGCCGGCCGTGTGATGAGACCAGTCTTTGTTGTCGAGCAGAGCACCACCGGGACCGTCGAGCAGGGCCATCTCGTCCTGCAAAAGGCCCTGGTCAACGAGATGCGCGAGCAACAGGAGGTGCCCCCTGACAACCCCGACGACAAGATCACCTGGCAGACGTTGGCCCACAGCGGTGTCATTGAGTATCTGGATGCCGAGGAAGAGGAGACGTCCATGATTTGCATGACGCCGGAGGACCTGGAGCTGTTCCGCAACCAAAAGGCCGGTTACGAATACCCCGACGACGTCGCAGAGGGTCCCAACAGGCGTCTGCGGACCAAGATTGCCAAGACGACGCACATGTACACGCATTGCGAGATTCACCCGAGCATGCTCCTCGGTATCTGCGCAAGCATTATTCCCTTCCCCGATCACAACCAGGTAAGTCTCCTGATACTGCTCTTGCGTGCCTTTGCTAACCCATGCCCGCGCAGTCTCCCCGTACAACTTACCG
>11532tub
GGGGCGTCCCGACGACGACGCCTCTCGACGAGACCAATCGACCACGGAATACTGACTTTTGGTCACAGGCAGAACATCTCTGGCGAGCACGGCCTCGACGCCAATGGCGTGTACAACGGCAGCTCCGAGCTCCAGCTCGAGCGCATGAACGTCTACTTCAACGAGGTATGCAGACCTCCACCCCCCCCCCCCCCCCCAAATCACTCCATCACTGCTAACCCGACGACAACAGGCCTCGAGCAACAAGTACGTTCCCCGTGCCGTCCTCGTCGATCTCGAGCCTGGTACCATGGACGCCATCCGCGCCGGTCCCTTCGGCCAGCTGTTCCGCCCCGACAACTTCGTCTCGGTCA
cf. Purpureocillium TUCIM11527
>11527ITS
GCGGAACGCGGAGGGATCTTACCGAGTTATACAACTCCCAAACCCACTGTGAACCTTACCTCAGTTGCCTCGGCGGGAACGCCCCGGCCGCCTGCCCCCGCGCCGGCGCCGGACCCAGGCGCCCGCCGCAGGGACCCCAAACTCTCTTGCATTACGCCCAGCGGGCGGAATTTCTTCTCTGAGTTGCACAAGCAAAAACAAATGAATCAAAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCGAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCGCCAGCATTCTGGCGGGCATGCCTGTTCGAGCGTCATTTCAACCCTCGAGCCCCCCCCGGGGGCCTCGGTGTTGGGGGACGGCACACCAGCCGCCCCCGAAATGCAGTGGCGACCCCGCCGCAGCCTCCCCTGCGTAGTAGCACACACCTCGCACCGGAGCGCGGAGGCGGTCACGCCGTAAAACGCCCAACTTTCTTAGAGTTGACCTCGGATCAGGTAGGAATACCCGCTGAACTTAAGCATATCAATAAGCGGGG
>11527rpb2
GGGGCTACTGGTGTGTCGCAGGTGCTCACCGTTACACATTTGCCTCGACCCTGTCGCATTTGCGGCGAACCAACACGCCCATCGGTCGTGACGGCAAGCTGGCCAAGCCCCGCCAGCTGCACAACACGCACTGGGGCCTCGTCTGTCCGGCTGAGACGCCAGAGGGACAGGCTTGCGGTCTGGTCAAGAATCTGTCTCTCATGTGCTACGTCAGTGTCGGCTCGCCGGCGGAGCCGCTCGTCGAGTTCATGATCAACCGAGGCATGGAGGTGGTGGAGGAGTACGAGCCGCTGAGATACCCCCACGCGACCAAGATCTTCGTCAACGGAGTCTGGGTTGGCGTGCACCAAGACCCCAAGCACCTGGTGAGCCAGGTGCTGGACACGCGGCGGAAGAGCTACCTCCCGTTCGAGGTGTCCCTCATCAGAGAGATCAGAGACCAAGAGTTCAAGGTCTTTTCCGACGCGGGACGAGTCATGCGCCCCGTCTTCACTGTGCAGCAGGAAGACGACCACGAGACGGGCGTGGGCAAGGGACAGCTGGTGTTGACCAAGGACCTCGTCAACAGGCTTGCCAAGGAGCAGTCCGAGCCGCCAGAGGACCCGAGCATGAAGCTGGGCTGGGAAGGCCTGATCAAGGCTGGTGCGGTGGAGTACCTGGACGCGGAGGAAGAGGAGACATCCATGATCTGCATGACGCCCGAGGACCTGGAGCTCTACCGCCTCCAGAAGGCCGGCGTTGCAGTGGACGATGACCCTGGCGATGACCTTAACAAGCGGCTGAAGACGAGGACCAACCCGACGACACACATGTACACCCACTGCGAGATTCACCCCAGCATGATCCTGGGCATCTGCGCCAGCATTATTCCCTTCCCGGATCATAACCAGGTATGTTTGCCCTCCCTGATGCCCCGGGCTCCGCCCTGACAGCTGAAGCCCCCCGAACCCTACAGTCTGTTGAA
>11527tef1
GTCGGGTGGCGGGAGCGTCTGTTCAGCACGTTAGCGACGTGGAAATTGCGACCAGTCTCGCGCGAGTCGACGTACCAATGACGGTGACATAGTACTTGGGAGTCTCGAACTTCCAGAGGGCAATGTCGATGGTGATACCACGCTCACGCTCGGCCTTGAGCTTGTCAAGGACCCACGCGTACTTGAAGGAACCCTTGCCGAGCTCGGCGGCTTCCTGTTATCTCACGCGTCAGCATGCTGTTGGTGCTCCGTGCGAATTTTTGGTGTTGAGGGGCAAATGAGACACCTGTGCAGCGTGGCGGGGTAATGAGCCCCACACAAAGCTGGCGACAAGTCACAGAAAATTTTGACTGTGCAAGGAGGGGTAATCGCCGCAATCAGAGCACATTGTGCACCGCTCGAGCGGCAGAACAGCTTGCTGCAAGGGAAAATGGAAAATCGAGTGAATCTTACCTTCTCGAACTTCTCGATGGTACGCTTGTCGATACCACCGCACTGGTAGATCAGGTGACCGGTCTGAGAGGTAGTTAGCATTCATGTCGCAGAAATACCCCGCGCCAGTGATGACAGGCGGGGCCTTGAACAGGGCGAGGAAGACTCACGGTGGTCGACTTGCCGGAGTCGACGTGGCTATTGCACGGAGTCAGTATGACGAGATCGCAGGCGACGACGATGGCATCGATTTACGCACCCCTGGAAGAGCGAGTTAGCAGACACATCTTTGAGGGAAAGTGAAAGTGGAAGAACGTACGATAACGACCACGTGATGGACTGCCCCCCCCTCTCTCACACACTAAACACCACACG
cf. Striaticonidium TUCIM11525
>11525ITS
GGCGTCGTAGGGACTTACCGAGTTTACAACTCCCAAACCCAATGTGAACATACCTAAACGTTGCCTCGGCGGGACCGCCCCGGCGCCCCTCGGGCCCGGAACCAGGCGCCCGCCGCAGGGAAACAAACCTCTGTTTTTGTTAAGATTTCTCCTCTGAGTGGATTTTACAAATAAATCAAAACTTTCAACAACGGATCTCTTGGCTCTGGCATCGATGAAGAACGCAGCGAAATGCGATACGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCGCCAGCATTCTGGCGGGCATGCCTGTTCGAGCGTCATTTCAACCCTCAGGCTCCCGCGCCTGGCGTTGGGGATCGGCCTTCACCGGCCGGCCCCGAAATACAGTGGCGGCCCCGCCCGTGTACCTCTGCGTAGTAGCACAACCTCGCAGCTGGGAGCGGCGGCGGCCACGCCGGAAAACCCCCGACTTCTGAAAGTTGACCTCGGATCAGGTAGGAATACCCGCTGAACTTAAGCATATCAATAAGCGGAGAAA
>11525rpb2
AACACTTAAACCCCTTTTGGGGTGGCCCCAAGAGAAGGCCATGAGTTCCACTGCTGGTGTTTCCCAGGTGCTCAACCGCTACACCTTCGCGTCCACTCTGTCGCATCTCCGGCGCACGAATACGCCCATTGGCCGAGACGGCAAGCTCGCAAAACCCCGCCAGCTGCACAACACGCACTGGGGTCTCGTCTGCCCCGCCGAGACGCCCGAAGGCCAAGCATGTGGTCTCGTGAAGAACTTGTCGTTGATGTGCTACGTCAGTGTCGGTTCCCGCGTTGATCCTCTCGTTGAATTCATGATTCAACGAGGAATGGAGGTGATTGAGGAGTACGAGCCCTTGAGATACCCCCATGCCACGAAGATTTTCGTCAATGGCGCTTGGATTGGTGTTCACCAAGACCCCAAGCACCTTGTCAACGCCGTCATGGACACGCGTCGTCGCTCTGTCATGCAATTCGAAGTCTCCCTTGTCCGAGACATTCGAGACCAAGAGTTCAAGATTTTTTCGGATGCCGGTCGTGTCATGCGACCAGTCTTCACTGTCTTGCAGGAAGACAACCCTGAGACGGGCCTTCAGAAGGGTCAGCTAGCGTTGACCAAAGAAATGGTCAACCAGCTTGCGGATGAGCAGTCAAACCCTATGCAAGACCCAGCAACGAAAATTGGCTGGCAGGGTCTAATCAAGAACGGTGCCGTGGAATATCTCGACGCCGAAGAAGAGGAGACAGCGATGATCTGCATGACTCCCGAGGATTTGGAGTTGTACAGGGCCCAGAAGGCTGGCAATCCGATCTTCGAGGACATGTCTGATGATCCCAACAAGAGACTGAAGACCAAGACCAATCCGACCACGCACATGTATACTCATTGCGAAATCCATCCTAGCATGATTCTGGGCATCTGCGCCAGTATCATTCCATTCCCCGATCACAACCAGGTAAGATCCCTCACTATATCTGAAGACGTGCCGGGCAACTTGATCGACTGATGCTCGTGTCCAGTCCCCGGTAACACTACCAGTGCTGTG
>11525cal
CCCCGGAGCGAGATGAACCGTTGTTATCGCGGTCAAAGACCTAGAAAGACGCGAGTCAGCAGGCGAAACGTCCAGATTAAAGACTGAAAACATACCTTGAAAGCTTCCCGAATCTCCTCCTCGGAGTCGGTGTCCTTCATCTTGCGTGCCATCATGGTGAGGAACTCTGCATCACACCCAAATCAGCCAACTGAAGGAGTGTAATTGCCGCTGGTTCGCCACCAGGTGCCACGGGGAGTGAGCCAACGTACCAGGGAAGTCGATGGTGCCGTTGTTGTCGGCGTCGACCTCGTTGATCATATCCTGCAGCTCAGACTCGGAGGGGTTCTGGCCCAGGGAGCGCATGACGGTTCCCAGCTCCTTGGTGGTGATCTGGCCTACAATGACGGGTAGGTTAGTGTCGGAGTCGAAGAAGCATTATCATCAACCCGGCAGCAAAGATGCTCCCAGAGTGAATACCATCGTTTTCGACCCGGATCGTGTGTTGCGGTCGCGCGTCGCAAGCGGTTTGTGAAGGGAGGAAGGGGGGGGGAGGGGGTAAGGCTGCCAGTCACTCACCATCGCCGTCCTTGTCCTATTTGCCCCGTTCGTCAGCATTGCATTGACAAGGAAGCTCGCAGCCACGGCGGGCTTGGTAGAAATGGGGAAAAGTAGCTTACAAAGAGGGAAAAGGCCCCTTTTTTAACCCCCGGAAAA
>11525tub
AAATATGCGTACGGACTGACCGAGACGAAGTTGTCGGGGCGGAAGAGCTGACCGAAGGGGCCAGCACGGACGGCGTCCATGGTACCGGGCTCGAGATCGACGAGGACGGCGCGAGGGACGTACTTGTCACCAGAGGCCTTTTATTGCAGCATGTCAGTGGTGCTGCTTGGATGGTTTGCCGCGCCTGGAGTTGGGGTTTTTTTCTTTTTTGGGGTATACCAACCTCGTTGAAGTAGACGCTCATGCGCTCGAGCTGAAGCTCGGAGGTGCCGTTGTACACACCATTGCTGTCGAGGCCGTGCTCGCCAGAGATGGTCTGCCAGAAAGCAGCACCATT
cf. Thyridium TUCIM11512
>11512ITS
CGATGCTACGCGGAGGGACATTAAAGAGTTGCAAAACTCCCAAACCATATGTTTATCTACCTGTTTCGTTGCTTCGGCAGGCGGTTCTTCTGGAGCCTTCGCCCTCTGGGTGCCTGCCGGAGTATCTAAAAACTCTTGTTAATTACTGTTCTGTCTGAGTAAACTTTTAATAAGTTAAAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCGAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCATTAGTATTCTAGTGGGCATGCCTGTTCGAGCGTCATTTCAACCCTCAAGCACTGCTTGGTGTTGGGGCGTCTGCAGCTCCGGCTGCAGGCCCTGAAAAACAGTGGCGGGCTCGCTATAACTCCGAGCGTAGTAATATATCCTCGCTTTGGAAGTATAGCGGTTCCCGGCCGTTAAACCCCCCAATTTCTGAACGTTGACCTCGGATCAGGTAGGAATACCCGCTGAACTTAAGCATATCAATAAGCGAGGAAAAAAAACCCCCCCCAAAAAAAGGAAGC
>11512rpb2
GAATAAGAAGCCGGTGTGTAAGGTGTTGACGATTACACCTTCGCATCCACCCTATCTCATTTGAGGCGTACCAACACTCCTATCGGTCGGGACGGAAAGCTGGCCAAGCCTCGCCAGCTGCACAACACCCACTGGGGTTTGGTCTGTCCAGCCGAGACGCCCGAGGGACAGGCTTGTGGTCTGGTCAAGAATTTGTCCTTGATGTGTTACGTCAGTGTGGGAACCCCGGCAGAGCCGATCGTGGAGTTCATGATTGCGAGAAACATGGAGGTCCTCGAGGAGTACGAGCCGCTGCGATACCCGAATGCCACCAAGATCTTTGTGAACGGTACATGGGTTGGTGTGCACCAGGATCCGAAACATCTTGTCACGCTGGTACAGGGATTACGACGGAAGGGAGTTATTAGCTTCGAGGTCTCTCTGGTCCGTGATATTCGCGACCGAGAGTTCAAGATCTTCTCTGATGCCGGCCGTGTCATGAGGCCGCTGTTTGCGGTCGAACAACAAGACGACGGCGAGAGCGGAGTCTCCAAGGGCTCCCTCATTCTCACCAAGCAGCACATCGCCAACCTCGAACAGGACCAGACATTGCCCAAGAGCGAGGCATGGGGATGGGACGGACTGGTTCACGCCGGCGCTATCGAGTATCTGGACGCCGAAGAGGAGGAGACGTCCATGATCTGCATGACGCCCGAGGACCTTGAGAACTACAAGCTGCAGAAGCAGGGTTACCAGCTACCGGACGAGATGGGCGACGGTGACCTCAACAAGCGTCTCAAGACCAAGATGAACCCGACAACGCACATGTACACGCACTGTGAGATCCACCCGAGCATGCTGCTAGGTATCTGCGCCAGCATCATCCCCTTCCCGGATCACAACCAGTCTCCCCGTACACCTCCCCCA
>11512tub
ACCGTGGGTTCTCTGCCGTTATGTCTGGCGTGACCACCTCTCTGCGCTTCCCCGGTCAGCTGAACTCTGACCTGCGCAAGCTGGCCGTCAACATGGTTCCCTTCCCCCGTCTCCACTTCTTCATGGTCGGCTTTGCTCCCCTGACCAGCCGTGGTGCCCACTCCTTCCGCGCCGTCAGCGTTCCCGAGCTGACCCAGCAAATGTTCGACCCCAAGAACATGATGGCTGCCTCTGACTTCCGCAACGGCCGCTACCTGACCTGCTCTGCCATCTTGTGAGTTTCCCAGTCTCTGAATCTCATCACTCGAGTAGCTAACAAGTCCTCTCCAGCCGCGGTAAGGTCTCCATGAAGGAGGTCGAGGACCAGATGCGCAACGTCCAGAACAAGAACTCATCCTACTTCGTTGAGTGGATCCCCAACAACGTCCAGACCGCTCTCTGCTCCATCCCCCCTCGTGGCCTCAAGATGTCTTCCACCTTTGTTGGAAACTCGACTGCCATCCAGGAGCTGTTCAAGCGTGTCGGCGAGCAGTTCACTGCCATGTTCAGGCGCAAGGCTTTCTTGCATTGGTATATGGGTGAGGGTAA
cf. Trichocladium TUCIM11529
>11529ITS
ATCGCGGAGGACATTAAGAGTTGCAAAACTCCCTAAACCATTGTGAACATACCTTCAACGTTGCTTCGGCGGGTTGGCTCCGGGTCTCCCGGGGCCCCCGGCCCTACTCGGGCGCCCGCCGGAGGTATCTAACTCTTGACAATTGTATGGCCTCTCTGAGTCTTTGTACTTAATAAGTCAAAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCGAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCGCCAGTATTCTGGCGGGCATGCCTGTTCGAGCGTCATTTCAACCATCAAGCCCCCGGCTTGTGTTGGGGACCTGCGGCTGCCGCAGGCCCTGAAAACCAGTGGCGGGCTCGCTAGTCACTCCGAGCGTAGTAATACATCTCGCTCAGGGCGTGCTGCGGGTTCCGGCCGTTAAAAAGCCTTATTTACCCAAGGTTGACCTCGGATCAGGTAGGAAGACCCGCTGAACTTAAGCATATCAATAAGCGAGGAAAAAAAACCCCCCCAAAAAAAAAAAAAAAAAAA
>11529rpb2
GGAGGAACTGCGCGGTGTGTCTAGGTGCTTAGCGCTACACATTTGCCTCAACGTTGTCACATTTGAGGCGGACCAACACGCCCATTGGTCGTGACGGCAAGCTTGCCAAACCCCGCCAGCTTCACAACACTCACTGGGGTCTCGTGTGCCCTGCCGAGACTCCCGAAGGACAGGCTTGCGGTCTCGTCAAGAACCTGTCCCTGATGTGCTACGTCAGCGTGGGTACGCCCGCGGATCCCATTATCGAATTCATGATCGCCAGAAACATGGAAGTGCTTGAAGAGTACGAGCCGCTCCGGTATCCCAATGCCACCAAGGTGTTTGTCAACGGCACCTGGGTCGGTGTTCATCAAGACCCGAAGCACCTGGTCTCGCTGGTCCAGGGCCTGCGGAGAAAGAACGTCATCTCGTTCGAGGTTTCGCTGGTTAGAGACATTCGCGACCGCGAGTTCAAGATCTTCTCCGATGCCGGGCGTGTCATGCGGCCGCTCTTCACGGTCGAACAGGAGGAGAACGGTGATAGCGGCGCTGAAAAGGGCCAGCTGATCCTCAACAAGGACCATATTCAACGGTTGGAGGCGGATAAGGAGCTCGGCAAATACCACCCCGACTATTGGGGCTGGCAGGGCCTGCTGCGGTCCGGCGCGATCGAGTACCTGGATGCTGAGGAGGAAGAGACAGTCATGATTTGCATGACGCCCGAAGACCTCGACATGTACCGCCTCACCAAGATGGGCTTCGACGTGGACGACACCTCTGGCCAAGGCAATAACCGGATCAGGACCAAGATGAACCCCACCACTCACATGTACACGCATTGTGAGATCCACCCCAGCATGCTGCTTGGTATCTGCGCGAGCATCATTCCTTCCCCGATCACAATCAGTCTCCCAGAAAGCCACC
cf. Trichoderma TUCIM11496
>11496ITS
GGTGACGCGTAGGGACATTACCGAGTTTACAACTCCCAAACCCAATGTGAACGTTACCAAACTGTTGCCTCGGCGGGATCTCTGCCCCGGGTGCGTCGCAGCCCCGGACCAAGGCGCCCGCCGGAGGACCAACCAAAACTCTTATTGTATACCCCCTCGCGGGTTTTTTTATAATCTGAGCCTTCTCGGCGCCTCTCGTAGGCGTTTCGAAAATGAATCAAAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCGAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCGCCAGTATTCTGGCGGGCATGCCTGTCCGAGCGTCATTTCAACCCTCGAACCCCTCCGGGGGGTCGGCGTTGGGGATCGGCCCTGCCTTGGCGGTGGCCGTCTCCGAAATACAGTGGCGGTCTCGCCGCAGCCTCTCCTGCGCAGTAGTTTGCACACTCGCATCGGGAGCGCGGCGCGTCCACAGCCGTTAAACACCCAACTTCTGAAATGTTGACCTCGGATCAGGTAGGAATACCCGCTGAACTTAAGCATATCAATAAGCGGAGGAAGAAAAACCCCAACCAAACCTTC
>11496rpb2
CATGACGATCTAGATCTACTTGGGTGATCTCGCAATGGGTGTACATGTGAGTTGTCGGGTTCGTCTTGGTCTTGAGTCGCTTGTTCGGATCATCTCCCATGTCTTCCTCAGTGTTGATACCGGCCTTCTGAAGACGATACAGCTCGAGATCCTCTGGTGTCATGCAGATCATGGCCGTCTCCTCTTCCTCGGCGTCGAGATATTCAACCGCACCAGCCCTGATCAATCCCTCCCATCCAATCTTCATGCTGGGGTCTTCCGGAGGCTCAGCCTGCTCCTTGGCCAATCTATTGACGAGCTCCTTGGTCAATACCAGGTGGCCCTTGTTGATGCCCGTTTCCGGATCATCTTCCTGCTGAACGGTAAAGACTGGTCGCATGACACGACCTGCATCGGAAAAGATTTTGAATTCCTGGTCTCGAATTTCTCTCACGAGAGAGACTTCGTATTGCAGATAGGACTTGCGACGAGTATCCAGAACCTGGTTCACCAAGTGCTTAGGGTCTTGGTGAACTCCAACCCAGACACCGTTCACAAAAATCTTTGTAGCATGAGGATACCGCAGCGGCTCGTACTCTTCGACGACTTCCATACCTCTGTTGATCATGAACTCAATCAGAGGCTCGGAGGGAGAACCGACACTGACGTAACACATCAAAGACAAGTTCTTGACCAGACCACAGGCCTGTCCTTCGGGGGTCTCGGCTGGGCAGACCAAGCCCCAATGCGTGTTGTGAAGCTGTCGAGGCTTCGCCAGCTTACCATCTCTCCCGATAGGAGTGTTGGTACGACGCAAATGTGACAAGGTCGAAGCAAACGTGTAACGGTTAAGCACCTGGGACACACCGGCAGTTGAGCTCATGGCCTTCTTCTGATCACCCCAGTTTCCTGTGGCAAGCGAATACTTCAGTCCGTTTGAAAGCGTGCCGGGCTTGATACCAACAGCCAGGTTGAAGTGTCGGTTGCCCTCAACGGATCGCTT
>11496tef1
GAGAAGGTAAGCTTCAACTGATTTTCGCCTCGATTCTTCCTCCTCCACATTCAATTGTGCCCGACAATTCTGCAGAGAATTTTCGTGTCGACAATTTTTCATCACCCCGCTTTCCATTACCCCTCCTTTGCAGCGACGCAAATTTTTTTTGCTGTCGTTTGGTTTTTAGTGGGGTTCTCTGTGCAACCCCACTAGCTTCCTGCTTTTTCCTGCTTCACTCTCACTTCCTCGTCATCATTCAACGTGCTCTGCGTCTTTGGTCATTCAGCGACGCTAACCACTTTTCCATCAATAGGAAGCCGCCGAACTCGGCAAGGGTTCCTTCAAGTACGCTTGGGTTCTTGACAAGCTCAAGGCCGAGCGTGAGCGTGGTATCACCATTGACATTGCTCTGTGGAAGTTCGAGACTCCCAAGTACTATGTCACCGTCATTGGTAAGTCTTCACTAAGTTCATGCTGCAATTGCGGACCAGTGCTAACAGGCAATTCACAGACGCTCCCGGCCACCG
cf. Trichoderma TUCIM11482
>11482ITS
GGTGACGCGGAGGGTCATTACCGAGTTTACAACTCCCAAACCCAATGTGAACGTTACCAAACTGTTGCCTCGGCGGGATCTCTGCCCCGGGTGCGTCGCAGCCCCGGACCAAGGCGCCCGCCGGAGGACCAACCAAAACTCTTTTTGTATACCCCCTCGCGGGTTTTTTTATAATCTGAGCCTTCTCGGCGCCTCTCGTAGGCGTTTCGAAAATGAATCAAAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCGAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCGCCAGTATTCTGGCGGGCATGCCTGTCCGAGCGTCATTTCAACCCTCGAACCCCTCCGGGGGGTCGGCGTTGGGGATCGGCCCTGCCTCTTGGCGGTGGCCGTCTCCGAAATACAGTGGCGGTCTCGCCGCAGCCTCTCCTGCGCAGTAGTTTGCACACTCGCATCGGGAGCGCGGCGCGTCCACAGCCGTTAAACACCCAACTTCTGAAATGTTGACCTCGGATCAGGTAGGAATACCCGCTGAACTTAAGCATATCAATAAGCGGAGGAAGAAAACACCCCCCCCAAAGGGTGGG
>11482rpb2
GACGATTGTAGGATGATACTTGGGTGATCTCGCAATGGGTGTACATGTGAGTTGTCGGGTTCGTCTTGGTCTTCAGTCGCTTGTTCGGATCATCTCCCATGTCTTCCTCAGTGTTAATACCGGCCTTCTGAAGACGATACAGCTCGAGATCCTCTGGCGTCATGCAGATCATGGACGTCTCCTCTTCCTCGGCGTCGAGATATTCAACTGCACCAGCCCTAATCAATCCCTCCCATCCAATCTTCATGCTGGGGTCCTCCGGAGGCTCAGCCTGCTCCTTGGCCAATCTATTGACGAGCTCCTTGGTTAATACCAGGTGGCCCTTGTTGATGCCCGTTTCCGGGTCATCTTCCTGCTGAACGGTAAAGACAGGTCGCATGACACGGCCTGCGTCGGAAAAGATTTTGAATTCCTGGTCTCGAATTTCTCTTACGAGAGAGACTTCGTATTGCAGATAGGACTTGCGACGAGTATCCAGGACCTGGTTCACCAAGTGCTTAGGGTCTTGGTGGACTCCAACCCAGACACCGTTCACAAAAATCTTTGTAGCATGAGGATACCTCAGCGGCTCGTACTCTTCGACAACTTCCATACCTCTGTTGATCATGAACTCAATCAGAGGTTCGGAGGGAGAACCGACACTGACGTAACACATCAAAGACAAGTTCTTGACCAGACCACAAGCCTGTCCTTCGGGTGTCTCGGCCGGGCAGACCAAACCCCAATGCGTGTTGTGAAGCTGTCGAGGCTTTGCCAGCTTACCATCTCTTCCGATGGGAGTGTTGGTACGACGCAAATGTGACAGGGTCGAAGCAAACGTGTATCGGTTAAGCACCTGGGACACACCTGCAGTTGAGCTCATGGCCTTCTTCTGATCACCCCAGTTTCCTGTGGCAAGCGAATACTTCAATCCGTTTGAAAGCGTGCCGGGCTTGATACCAACAGCAAGGTTGAAGTGTCGGTTGCCCTCACCCATCGTCTCAATAGTGGCATCGTCAATTTT
>11482tef1
GAGAAGGTAAGCGTCAACTGATTTTCGCCTCGATTCCTCTTCTTTCATATTCAATTGTGCCCGACAATTCTTCAAAGACTTTTTGGGTCGACAATTTTTCGTCACCCCGCTTTCCATTACCCCTCCTTTGCAGCGACGCAAATTTTTTTTGCTGCCGTTTGATTTTTAGTGGGGTTCTCTGTGCAACCCCACTAGCTCACTGCTTTTTTTGTGCTTCATTCACTTCCCAGTCATCATTCAACGTGCTCTGTGTCTTTGGTTATTCAACGATGCTAACCACTTTTCCATCAATAGGAAGCCGCCGAACTCGGCAAGGGTTCCTTCAAGTACGCTTGGGTTCTTGACAAGCTCAAGGCCGAGCGTGAGCGTGGTATCACCATCGACATTGCTCTGTGGAAGTTCGAGACTCCCAAGTACTATGTCACCGTCATTGGTATGTCTACTTCATCAACTTCATGCTGCAATTGCAACCCAGTGCTAACAGGCAATTCACAGACGCTCCCGGCCACCG
cf. Trichoderma TUCIM11470
>11470ITS
GGGTGACGCGGAGGGACATTACCGAGTTTACAACTCCCAAACCCAATGTGAACGTTACCAAACTGTTGCCTCGGCGGGATCTCTGCCCCGGGTGCGTCGCAGCCCCGGACCAAGGCGCCCGCCGGAGGACCAACCTAAAACTCTTTTTGTATACCCCCTCGCGGGTTTTTTTATAATCTGAGCCTTCTCGGCGCCTCTCGTAGGCGTTTCGAAAATGAATCAAAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCGAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCGCCAGTATTCTGGCGGGCATGCCTGTCCGAGCGTCATTTCAACCCTCGAACCCCTCCGGGGGGTCGGCGTTGGGGATCGGCCCTGCCTCTTGGCGGTGGCCGTCTCCGAAATACAGTGGCGGTCTCGCCGCAGCCTCTCCTGCGCAGTAGTTTGCACACTCGCATCGGGAGCGCGGCGCGTCCACAGCCGTTAAACACCCAACTTCTGAAATGTTGACCTCGGATCAGGTAGGAATACCCGCTGAACTTAAGCATATCAATAAGCGGAGGAAGAAACACCCCCCCCCAAAGGT
>11470rpb2
GGCAAAAACAGACCCGTTAGGATAATACGTGGGGAAGATCTCGCAGTGGGTGTACATGTGAGTTGTGGGGTTTGTCTTTGTCTTCAGTCGCTTGTTCGGATCATCTCCCATGTCTTCCTCAGTGTTAATACCGGCCTTCTGAAGACGATACAGCTCGAGATCCTCTGGCGTCATGCAGATCATGGACGTTTCCTCTTCCTCGGCGTCGAGATATTCAACTGCACCAGCCCTAATCAATCCCTCCCATCCAATCTTCATGCTGGGGTCTTCCGGAGGCTCAGCCTGCTCCTTGGCCAATCTATTGACGAGTTCCTTGGTCAATACCAGGTGGCCCTTGTTGATGCCCGTTTCCGGGTCATCTTCCTGCTGAACGGTAAAGACTGGTCGCATGACACGACCAGCGTCGGAAAAGATTTTGAATTCCTGGTCTCGAATTTCTCTCACAAGAGAGACTTCGTACTGCAGATAGGACTTGCGACGAGTGTCCAGGACCTGGTTCACCAAGTGCTTAGGGTCTTGGTGAACTCCAACCCAGACACCGTTCACAAAAATCTTTGTAGCATGAGGATACCGCAGCGGCTCGTACTCTTCGACGACTTCCATGCCTCTGTTGATCATGAACTCAATCAGAGGCTCAGAGGGAGAACCGACACTGACGTAACACATCAAAGACAAGTTCTTGACCAGACCACAGGCCTGTCCTTCGGGTGTTTCGGCTGGGCAGACCAAACCCCAATGCGTGTTGTGAAGCTGTCGAGGCTTTGCCAGCTTACCATCTCTTCCGATGGGAGTGTTGGTACGACGCAAATGTGACAGGGTCGAAGCAAACGTGTATCGGTTAAGCACCTGGGACACACCTGCAGTTGAGCTCATGGCCTTCTTCTGATCACCCCAGTTGCCTGTGGCAAGCGAATACTTCAATCCGTTTGAAAGCGTGCCGGGCTTGATACCAACAGCAAAGGTTGAAGTGTCGGTTGCCCTCAACGCATCGTCTCAGATAGTTGGGCAATTCATGCTCCCCCTCTCTCGCGCATATATAA
>11470tef1
GAGAAGGTAAGCTTCAACTGATTTTTCGCCTCGATTCCTCTCATATTCAATTGTGCCCGACAATTCTTCAGAGAATTTTCGTGTCGACAATTTTTCATCACCCCGCTTTCCATTACCCCTCCTTTGCAGCGACGCAAATTTTTTTTGGTGCCGTTTGATTTTTAGTGGGGTTCTCTGTGCAACCCCACTAGCTCACTGCTTTTTTTTTTGTGCTTCACTCACTTCCCAGTCATCATTCAACGTGCTCTGTGTCTTTGGTCATTCAACGATGCTAACCACTTTTCCATCAATAGGAAGCCGCCGAACTCGGCAAGGGTTCCTTCAAGTACGCTTGGGTTCTTGACAAGCTCAAGGCCGAGCGTGAGCGTGGTATCACCATCGACATTGCTCTGTGGAAGTTCGAGACTCCCAAGTACTATGTCACCGTCATTGGTATGTCTACTTCACCAACTTCATGCTGCAATTGCAACCCAGTGCTAACAGGCAATTCACAGACGCTCCCGGCCACCG
cf. Trichoderma TUCIM11491
>11491ITS
TTGGTGACGCGGAGGGACATTACCGAGTTTACAACTCCCAAACCCAATGTGAACGTTACCAAACTGTTGCCTCGGCGGGATCTCTGCCCCGGGTGCGTCGCAGCCCCGGACCAAGGCGCCCGCCGGAGGACCAACCTAAAACTCTTTTTGTATACCCCCTCGCGGGTTTTTTTATAATCTGAGCCTTCTCGGCGCCTCTCGTAGGCGTTTCGAAAATGAATCAAAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCGAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCGCCAGTATTCTGGCGGGCATGCCTGTCCGAGCGTCATTTCAACCCTCGAACCCCTCCGGGGGGTCGGCGTTGGGGATCGGCCCTGCCTCTTGGCGGTGGCCGTCTCCGAAATACAGTGGCGGTCTCGCCGCAGCCTCTCCTGCGCAGTAGTTTGCACACTCGCATCGGGAGCGCGGCGCGTCCACAGCCGTTAAACACCCAACTTCTGAAATGTTGACCTCGGATCAGGTAGGAATACCCGCTGAACTTAAGCATATCAATAAGCGGAGGAAGAAAAACCCCCACCAAAGGTTCGGT
>11491rpb2
GAGAGGCCTAGGATGTACTTGGGTGATCTCGCAATGGGTGTACATGTGAGTTGTCGGGTTCGTCTTGGTCTTCAGTCGCTTGTTCGGATCATCTCCCATGTCTTCCTCAGTGTTGATACCGGCCTTCTGAAGACGATACAGCTCGAGATCCTCTGGCGTCATGCAGATCATAGACGTCTCCTCTTCCTCGGCGTCGAGATATTCAACCGCACCAGCCCGAATCAATCCCTCCCATCCAATCTTCATGCTGGGGTCTTCCGGAGGCTCAGCCTGCTCCTTGGCCAATCTATTGACGAGTTCCTTGGTCAATACCAGGTGGCCCTTGTTGATGCCCGTTTCCGGGTCATCTTCCTGCTGAACGGTAAAGACTGGTCGCATGACACGACCAGCGTCGGAAAAGATTTTGAATTCCTGGTCTCGAATTTCTCTCACAAGAGAGACTTCGTACTGCAGATAGGACTTGCGACGAGTGTCCAGGACCTGGTTCACCAAGTGCTTAGGGTCTTGGTGAACTCCAACCCAGACACCGTTCACAAAAATCTTTGTAGCATGAGGATACCGCAGCGGCTCGTACTCTTCGACGACTTCCATGCCTCTGTTGATCATGAACTCAATCAGAGGCTCAGAGGGAGAACCGACACTGACGTAACACATCAAAGACAAGTTCTTGACCAGACCACAGGCCTGTCCTTCGGGTGTTTCGGCTGGGCAGACCAAACCCCAATGCGTGTTGTGAAGCTGTCGAGGCTTTGCCAGCTTACCATCTCTTCCGATGGGAGTGTTGGTACGACGCAAATGTGACAGGGTCGAAGCAAACGTGTATCGGTTAAGCACCTGGGACACACCTGCAGTTGAGCTCATGGCCTTCTTCTGATCACCCCAGTTGCCTGTGGCAAGCGAATACTTCAATCCGTTTGAAAGCGTGCCGGGCTTGATACCACAGCAAGGTTGAAGTGCCGGGTGCCTC
>11491tef1
GAGAAGGTAAGCTTCAACTGATTTTTCGCCTCGATTCCTCTCATATTCAATTGTGCCCGACAATTCTTCAAAGAATTTTCGTGTCGACAATTTTTCATCACCCCGCTTTCCATTACCCCTCCTTTGCAGCGACGCAAATTTTTTTTGGTGCCGTTTGATTTTTAGTGGGGTTCTCTGTGCAACCCCACTAGCTCACTGCTTTTTTTTTTGTGCTTCACTCACTTCCCAGTCATCATTCAACGTGCTCTGTGTCTTTGGTCATTCAACGATGCTAACCACTTTTCCATCAATAGGAAGCCGCCGAACTCGGCAAGGGTTCCTTCAAGTACGCTTGGGTTCTTGACAAGCTCAAGGCCGAGCGTGAGCGTGGTATCACCATCGACATTGCTCTGTGGAAGTTCGAGACTCCCAAGTACTATGTCACCGTCATTGGTATGTCTACTTCACCAACTTCATGCTGCAATTGCAACCCAGTGCTAACAGGCAATTCACAGACGCTCCCGGCCACCG
cf. Trichoderma TUCIM11471
>11471ITS
GGGTACGCGGAGGGACATTACCGAGTTTACAACTCCCAAACCCAATGTGAACGTTACCAAACTGTTGCCTCGGCGGGATCTCTGCCCCGGGTGCGTCGCAGCCCCGGACCAAGGCGCCCGCCGGAGGACCAACCAAAACTCTTATTGTATACCCCCTCGCGGGTTTTTTTATAATCTGAGCCTTCTCGGCGCCTCTCGTAGGCGTTTCGAAAATGAATCAAAACTTTCAACAACGGATCTCTTGGTTCTGGCATCGATGAAGAACGCAGCGAAATGCGATAAGTAATGTGAATTGCAGAATTCAGTGAATCATCGAATCTTTGAACGCACATTGCGCCCGCCAGTATTCTGGCGGGCATGCCTGTCCGAGCGTCATTTCAACCCTCGAACCCCTCCGGGGGGTCGGCGTTGGGGATCGGCCCTGCCTCTTGGCGGTGGCCGTCTCCGAAATACAGTGGCGGTCTCGCCGCAGCCTCTCCTGCGCAGTAGTTTGCACACTCGCATCGGGAGCGCGGCGCGTCCACAGCCGTTAAACACCCAACTTCTGAAATGTTGACCTCGGATCAGGTAGGAATACCCGCTGAACTTAAGCATATCAATAAGCGGAGGAAGAAAACACCCCCCCCCAGGGGGCGGGT
>11471rpb2
AAACGGGAACCCTAGATCATACTTGGGTGATCTCGCAGTGGGTGTACATGTGAGTTGTCGGGTTCGTCTTGGTCTTCAGTCGCTTGTTCGGATCATCTCCCATGTCTTCCTCAGTGTTAATACCGGCCTTTTGAAGACGATACAGCTCGAGATCCTCTGGCGTCATGCAGATCATGGACGTCTCCTCTTCCTCGGCGTCGAGATATTCAACTGCACCAGCCCTAATCAATCCCTCCCATCCAATCTTCATGCTGGGGTCTTCCGGAGGCTCAGCCTGCTCCTTGGCCAGTCTATTGACGAGCTCCTTGGTCAATACCAGGTGGCCCTTGTTGATGCCCGTTTCCGGGTCATCTTCCTGCTGAACGGTAAAGACTGGTCGCATGACACGACCTGCGTCGGAAAAGATTTTGAATTCCTGGTCTCGAATTTCTCTCACGAGAGAGACTTCGTATTGCAGATAGGACTTGCGACGAGTATCTAGAACCTGGTTCACCAAGTGCTTAGGGTCTTGGTGAACTCCAACCCAGACACCGTTCACGAAAATCTTTGTAGCATGAGGATACCGCAGCGGCTCGTACTCTTCGACGACTTCCATACCTCTGTTGATCATGAACTCAATCAGAGGCTCAGAAGGAGAACCGACACTGACGTAACACATCAAAGACAAGTTCTTGACCAGACCACAGGCCTGTCCTTCGGGTGTCTCGGCTGGGCAGACCAAACCCCAATGCGTGTTGTGAAGTTGTCGAGGCTTTGCCAGCTTACCATCTCTTCCGATGGGAGTGTTGGTACGACGCAAATGTGACAGGGTCGAAGCAAACGTATATCGGTTAAGCACCTGTGACACACCTGCCGTTGAGCTCATGGGCCTTCTTCTGATCACCCCAGTTGCCTGTGGCAAGCGAATACTTCAATCCGTTTGAAAGCGTGCCGGGCTTGATACCAACAGCAAGGTTGAAGTGTCGGTTGCCCTCAACGCATCGCCTCAGGTAGTTGGGCAACTCAGTACTCCCCCCTCTCGCGCCTCTAGAAG
>11471tef1
GAGAAGGTAAGCTTCAACTCATTTTCGCCTCGATTCTCCCTCCACATTCAATTGTCCCCGACAATTCTGCAGAGAATTTTCGTGTCGACAATTTTTCATCACCCCGCTTTGCATTACCCCTCCTTTGCAGCGACGCAAATTTTTTTTGCTGTCGTTTGGTTTTAGTGGGGTTTCTTGTGCACCCCACTAGCTCATGGCTTTTTTTCTGCTTTGCTCTCACGTCGCAGCCATCATTCAACGTGCTCTGTGTCTCGTCACTTTCAGCGATGCTAACCACTTTTCCATCAATAGGAAGCCGCCGAACTCGGCAAGGGTTCCTTCAAGTACGCTTGGGTTCTTGACAAGCTCAAGGCCGAGCGTGAGCGTGGTATCACCATCGACATTGCTCTGTGGAAGTTCGAGACTCCCAAGTACTATGTCACCGTCATTGGTATGTTCTTTCCATCAACTTCACACAGTGATTACAAGCCAGTGCTAACAAGCGATTTATAGACGCTCCCGGCCACCG